多種碳正離子的結構和穩定性問題的探討
2024-01-23 12:55:02王文峰袁耀鋒
大學化學 2023年11期
王文峰,袁耀鋒
福州大學化學學院,福州 350116
1 引言
碳正離子是《有機化學》上冊教學中最重要的中間體,其結構和穩定性一直是物理有機化學家最感興趣的研究內容[1,2]。由于課時限制,多數高校在碳正離子的教學中只能介紹一些已有定論的簡單知識,不能滿足今后有志于從事有機化學研究工作的學生的需求。本文計算了多種碳正離子的結構和穩定性問題,旨在使大學生對這個中間體有一個更深入的了解。
2 計算方法
本文采用Gaussian 03[3]程序和密度泛函理論(DFT)中的B3LYP方法計算所有化合物的分子結構,對所有原子采用6-311+G**基組并用全優化方法計算其穩定結構。
3 碳正離子結構和穩定性
3.1 碳正離子的超共價結構
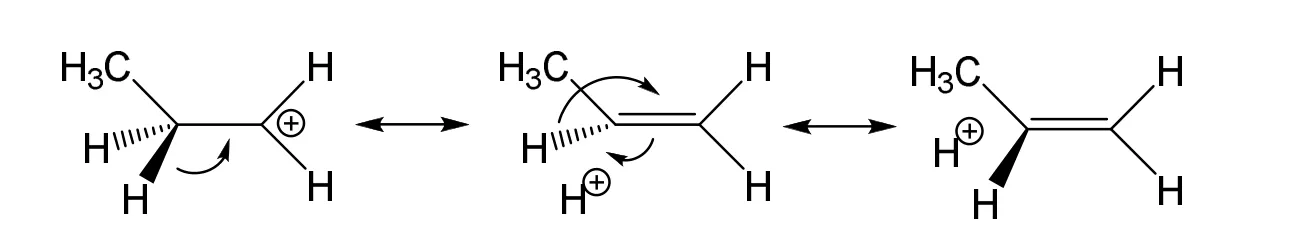
許多權威的有機化學教科書[4,5]都介紹碳正離子為sp2雜化,碳形成三個共平面的共價鍵。絕大部分碳正離子的結構的確如此,但是有例外情況。物理有機化學家的研究證實[6]51-52(中括號外為頁碼,后同),乙基碳正離子的5個H原子無論在溶液中還是在氣相中都以很快的速率進行重排。這種現象表明碳正離子很可能具有如Scheme 1所示的動態結構,但是在動態結構中,橋形結構和開環結構哪一個具有更低的能量,物理有機化學家不能確定。
本文采用DFT方法對乙基碳正離子的結構和穩定性進行了計算。DFT方法優化乙基碳正離子的結構時,依據初始結構的不同,可以得到兩個不同的乙基碳正離子結構。第一種初始結構為:甲基中有一個C—H與碳正離子所在平面共平面,如圖1A所示,C1—H3鍵與碳正離子所在平面共平面,同時這個平面均勻分開C1—H4和C1—H5兩個共價鍵。在這種結構中,沒有一個H處于可遷移的位置,所以本文稱之為不遷移初始結構。第二種初始結構為:甲基中有一個C—H鍵與碳正離子的未雜化p軌道共平面,如圖1B的C1—H3鍵,這種結構就是Scheme 1中的開環結構。這種構型中C1—H3鍵可以和碳正離子發生超共軛效應,且H3原子可以遷移到鄰近的碳正離子上,本文稱之為可遷移初始構型。這兩種初始構型優化后的結構分別顯示于圖1C和圖1D。

圖1 乙基碳正離子的初始構型和優化后結構
從不遷移初始構型的優化后結構圖1C來看,優化后的確沒有H原子發生遷移,碳正離子也的確是平面型的,因為二面角H2—C2—H1—C1為179.9°。甲基中的三個C—H鍵長有差別,C1—H3鍵因為與碳正離子共平面,與碳正離子完全沒有超共軛效應,所以鍵長只有0.1086 nm。C1—H4和C1—H5不與碳正離子共平面,有部分超共軛效應,鍵長被部分拉長,分別為0.1118和0.1117 nm。
可遷移初始構型優化后H3原子的確發生了遷移,得到了橋形結構圖1D。在圖1D中,C—C鍵長只有0.1381 nm,兩個碳原子和四個氫原子非常接近共平面,同時H3原子與兩個碳原子的鍵長都在0.1322 nm左右,比正常C—H鍵長得多。仿佛一個H+浮在一個平面乙烯分子上方。這種結構中H3原子同時形成了兩個共價鍵,與Scheme 1中的橋型結構相同,這種H原子被稱為超共價原子[6]51-52,本文將這種含有超共價原子的結構稱為超共價結構(Super covalent structure)。以超共價結構圖1D的能量為零,則不遷移初始構型優化后結構圖1C的能量為15.7 kJ·mol-1。不遷移結構和可遷移結構都屬于開環結構,可遷移結構優化后得到的是橋型結構(即超共價結構),而不遷移結構優化后能量高于橋型結構,這些結果說明Scheme 1中橋型結構比開環結構更穩定。
3.2 丙基碳正離子的結構
是不是所有碳正離子的最穩定結構都是超共價結構呢?為回答這個問題,本文選擇對丙基碳正離子進行計算。采用類似圖1的方式,給出兩種初始構型分別進行優化計算,結果如圖2所示。
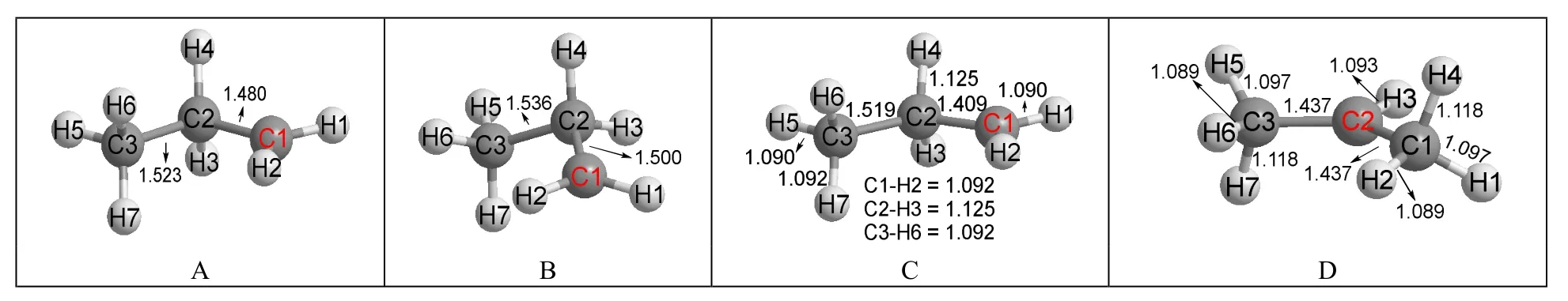
圖2 丙基碳正離子的初始構型和優化后結構
在圖2中,兩個初始構型(圖2A和圖2B)顯示的都是正丙基碳正離子的結構,即。與圖1的計算結果類似,不遷移初始構型優化后沒有發生H原子的遷移,得到的仍然是正丙基碳正離子的結構(圖2C)。圖2C結構中C2—H4和C2—H3的鍵長均為0.1125 nm,比其余C—H鍵更長,表明這兩個C—H鍵與相鄰的碳正離子存在超共軛效應。另外,圖2C結構中C2—C1的鍵長(0.1409 nm)比C2—C3的鍵長(0.1519 nm)短得多,原因是超共軛效應的存在使得C2—C1鍵具有雙鍵性質,如Scheme 2所示:
Scheme 2 丙基碳正離子通過無鍵共振形成超共軛效應的結構表示圖
在可遷移初始構型優化后的結構圖2D中,H4原子的確發生了遷移,得到的是異丙基碳正離子的結構,即CH3CH+CH3。以圖2D結構的能量為參照(即設為0.0 kJ·mol-1),則圖2C結構的相對能量是61.6 kJ·mol-1。這個結果表明仲碳正離子比伯碳正離子穩定得多,該結論與大學教科書結論一致。
乙基碳正離子之所以采用超共價結構,一個重要原因是它的兩個經典結構是等價的。但丙基碳正離子的兩個經典結構分別是正丙基碳正離子和異丙基碳正離子(以H原子為遷移原子),能量相差很大,完全不等價,所以其最穩定結構是異丙基碳正離子而不是超共價結構。異丙基碳正離子具有對稱結構(見圖2D),左右兩個甲基是等價的,所以其結構中顯示了等價的超共軛效應:C3—H7鍵和C1—H4鍵都和碳正離子未雜化的p軌道共平面,它們與碳正離子的超共軛效應最強,鍵長都是0.1118 nm,在所有C—H鍵中最長;C3—H6鍵和C1—H2鍵由于與碳正離子所在平面幾乎共平面,這兩個C—H鍵幾乎沒有與碳正離子發生超共軛作用,它們的鍵長0.1089 nm是所有C—H鍵中最短的。C3—H5和C1—H1則由于與碳正離子存在程度很小的超共軛作用,所以它們的鍵長值0.1097 nm處于中間。此外,C2正離子和C1及C3兩原子的鍵長都是0.1437 nm,而叔丁基碳正離子[(CH3)3C+]的晶體結構中碳碳鍵長是0.1442 nm[6]50,兩者十分接近,表明本文的計算結果可靠。
3.3 仲丁基碳正離子的結構
丙基碳正離子因為具有不等價的經典結構而不采取超共價結構,那么是不是具有等價經典結構的碳正離子都會像乙基碳正離子那樣以超共價結構作為最穩定結構呢?為了回答這個問題,本文選擇對仲丁基碳正離子進行結構優化。因為仲丁基碳正離子的兩個經典結構是等價的,如Scheme 3所示。本文以可遷移構型為初始構型,考察H原子是否會遷移形成超共價結構。計算結果列在圖3。
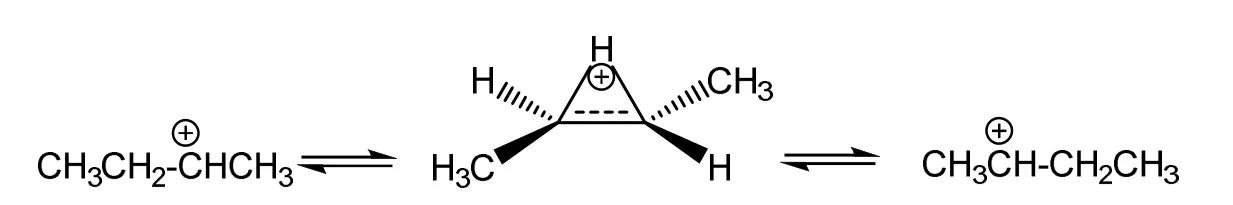
Scheme 3 仲丁基碳正離子的超共價結構和經典結構
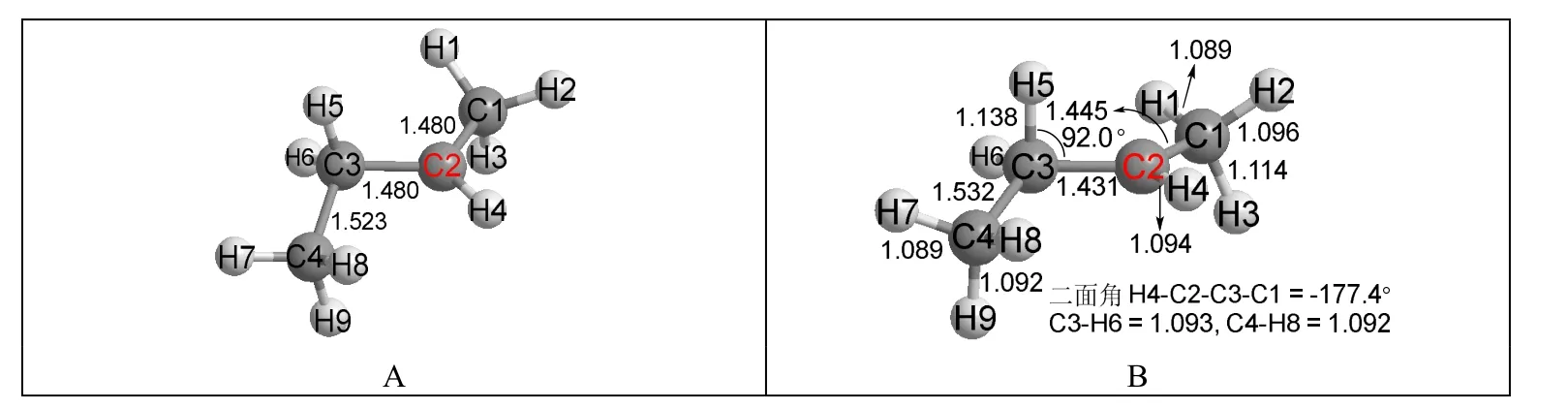
圖3 仲丁基碳正離子的結構
在圖3A中,H3和H5原子都處在可遷移的位置。但是經過優化后,這兩個原子都沒有發生遷移。而且仲丁基碳正離子與異丙基碳正離子不同,它不是對稱結構,C2正離子與相鄰的CH3和CH2上的C—H鍵的超共軛效應不等價,此時如何選擇?從圖3B的結構中能發現如下幾點:(1) C2—C3的鍵長0.1431 nm短于C2—C1的鍵長0.1445 nm,表明C2—C3比C2—C1具有更多的雙鍵成分。原因是C2—C1形成的雙鍵是末端雙鍵,超共軛效應弱,而C2—C3雙鍵是鏈內雙鍵,超共軛效應強;(2) C3—H5的鍵長0.1138 nm比C1—H3的鍵長0.1114 nm長,表明C3—H5與碳正離子的超共軛效應比C1—H3強;(3) H5—C3—C2的鍵角只有92.0°,遠遠偏離了sp3雜化碳原子的正常鍵角109.5°,是因為接近90°的鍵角能和碳正離子的空p軌道更好地發生重疊;而C2—C1—H3的鍵角是103.0° (圖3中未列出),進一步表明C3—H5的超共軛效應強于C1—H3的超共軛效應。這些結果指向一個結論:由于形成雙鍵是發生超共軛作用的附帶結果(如Scheme 2所示),所以碳正離子總是傾向于與能形成更穩定雙鍵的碳原子上的C—H鍵發生超共軛作用,而且這種超共軛作用能較大程度改變C—H的鍵長和鍵角。
3.4 仲碳正離子到叔碳正離子的重排
3.2部分的計算表明伯碳正離子優化后會得到仲碳正離子,那么仲碳正離子優化后是否會得到叔碳正離子?本文以可遷移構型為初始構型,對3-甲基-2-丁基碳正離子進行了優化,結果見圖4。

圖4 3-甲基-2-丁基碳正離子的結構
由圖4可見,優化后H5原子確實遷移到C2原子上,C3成為一個叔碳正離子。這表明仲碳正離子在優化時也會重排成更穩定的叔碳正離子。結合3.2部分的計算,可以得出一個推論:如果一個碳正離子能夠重排成更穩定的碳正離子,則這種重排反應非常容易發生。
3.5 以碳為超共價原子的碳正離子
在前述的所有討論中,本文都以H原子作為遷移基團。如果以CH3作為遷移基團,能不能獲得以碳為超共價原子的碳正離子呢?如果能,這樣的碳正離子是否能比以H原子為超共價原子的碳正離子穩定?為了回答這個問題,本文選擇將CH3作為遷移基團來優化正丙基碳正離子CH3CH2CH2+的結構。結果列于圖5。
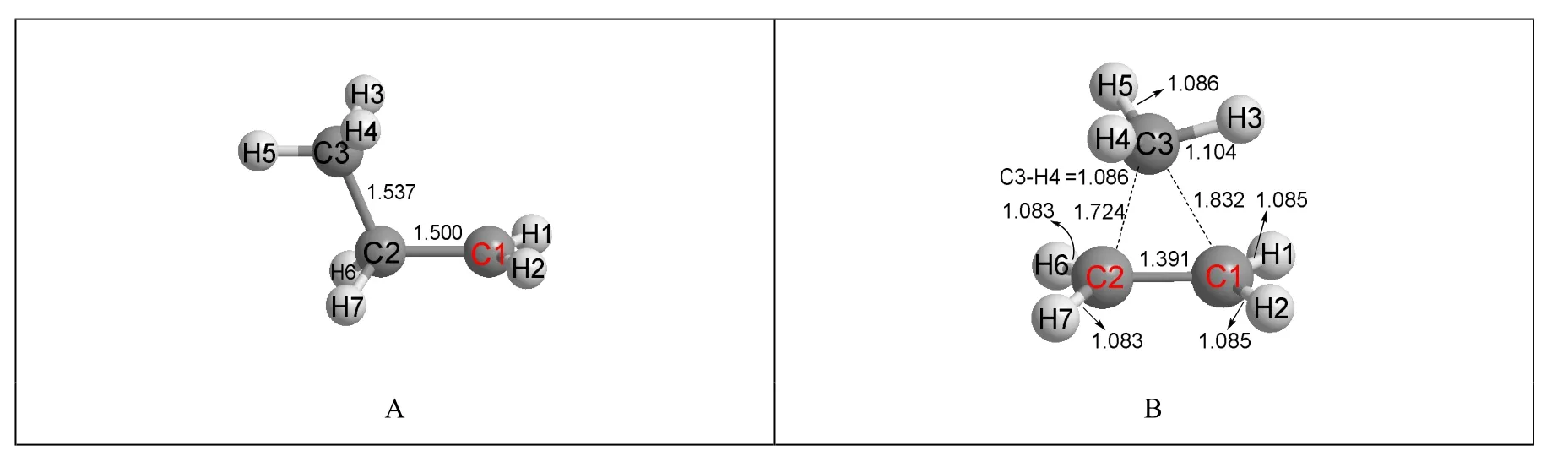
圖5 以甲基為遷移基團的丙基碳正離子的結構
在優化后的結構圖5B中,甲基確實發生了遷移,形成了一個以碳原子為超共價原子的超共價結構。但與乙基超共價結構圖1D有區別的是,下方的兩個碳原子(C1和C2)與四個氫原子(H1、H2、H6和H7)不共平面,且C3原子不處于C1和C2兩個原子的中間位置,即C3—C2與C3—C1的鍵長不相等。但整個分子有對稱面,C1、C2、C3與H3這四個原子共平面,且將分子中剩余的6個氫原子平分在平面兩端,所以該平面即分子的對稱面。與H作為遷移基團的優化后結構圖2D的能量相比,若圖2D的能量為0.0,則圖5B的能量為44.15 kJ·mol-1,遠遠不如圖2D穩定。這說明以碳為超共價原子的超共價結構不是碳正離子的穩定結構,同時也表明以氫原子作為可遷移基團來進行碳正離子結構優化的方法是正確的。
3.6 烯丙型碳正離子的穩定性問題
在基礎有機化學教科書中,烯丙型碳正離子和叔碳正離子都屬于非常穩定的碳正離子,但沒有介紹哪一種碳正離子更穩定。在高等有機化學教科書中,用氫負離子的親和性(定義為氫負離子與碳正離子結合為相應烷烴所放出的能量,HIA)來衡量氣態碳正離子的穩定性[6]85-86,表1列出一些氣態碳正離子的HIA值。

表1 丁基碳正離子與烯丙基型碳正離子和芐基碳正離子的HIA值
HIA值越小,碳正離子越穩定。表1列出的三個烯丙基碳正離子都是伯烯丙基碳正離子。沒有取代基的烯丙基碳正離子的HIA是1070 kJ·mol-1,穩定性還不如仲碳正離子。有一個和兩個取代基的伯烯丙基碳正離子的HIA分別是986和940 kJ·mol-1,已經分別比仲碳正離子(1032 kJ·mol-1)和叔碳正離子(966 kJ·mol-1)穩定。這表明烯丙型碳正離子隨取代基的不同,穩定性有很大差別。
本文優化計算了如圖6所示的五種同分異構體,依據碳正離子和雙鍵位置的不同,它們分別屬于仲碳正離子、叔碳正離子、一取代伯烯丙型碳正離子、仲烯丙型碳正離子和叔烯丙型碳正離子。
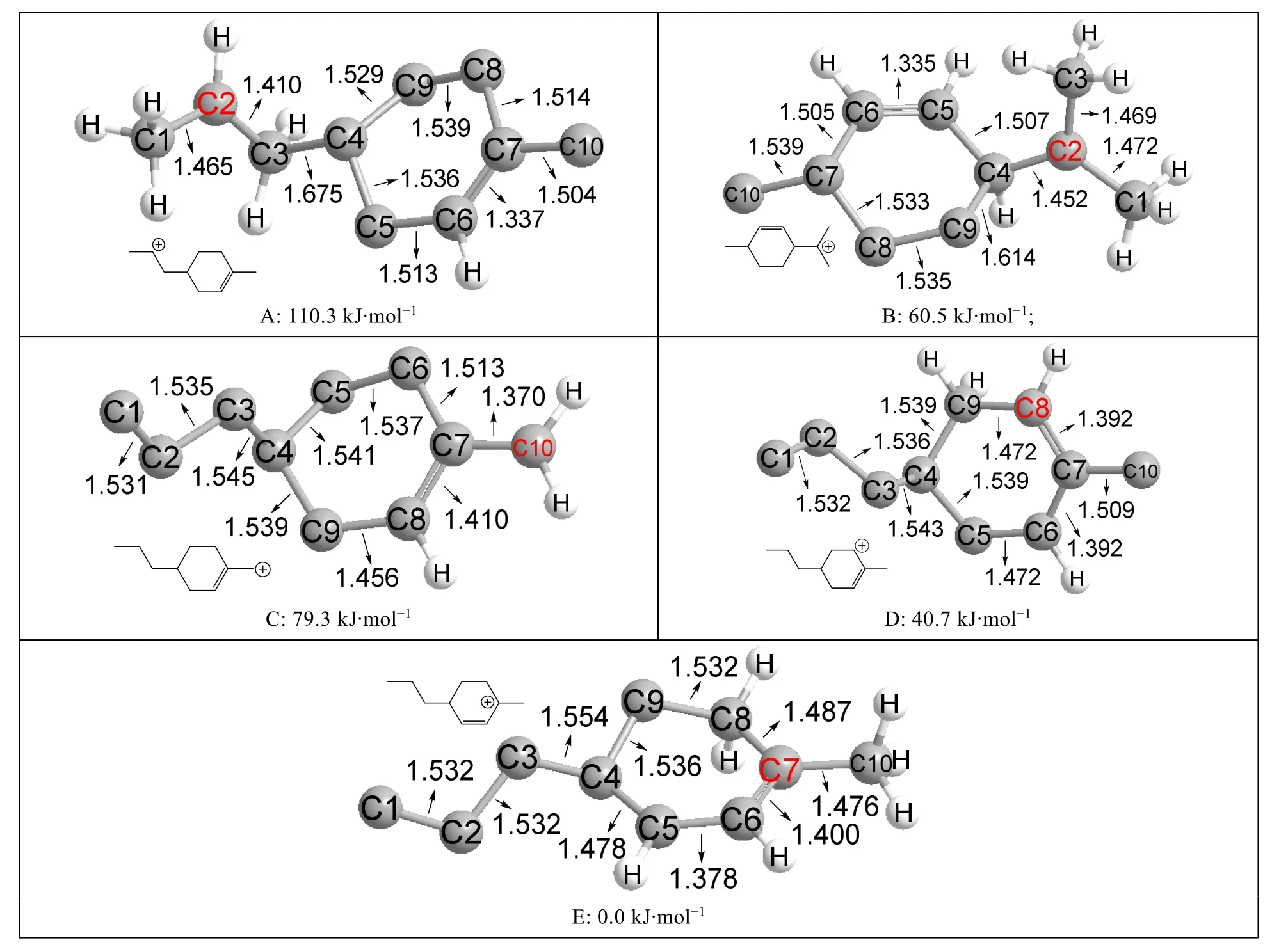
圖6 五種碳正離子同分異構體優化后的結構圖及其相對能量
以最穩定的叔烯丙型碳正離子為參照,其能量設為0.0 kJ·mol-1。從圖6中可以看出,叔碳正離子的穩定性比仲碳正離子穩定得多,能量差約50 kJ·mol-1;一取代伯烯丙基碳正離子比仲碳正離子穩定,但不如叔碳正離子穩定,與表1的計算結果相同。仲烯丙型碳正離子已經比叔碳正離子穩定,叔烯丙型碳正離子則比仲烯丙型碳正離子更穩定,兩者能量差約40 kJ·mol-1。
在圖6D的結構中,C7—C6和C7—C8的鍵長都是0.1392 nm,表明碳正離子和雙鍵已經完全離域。類似地,圖6C和圖6E的結構中碳正離子和雙鍵也存在明顯離域。所以烯丙型碳正離子的穩定性可以歸因于共軛效應。
3.7 一些特別穩定的碳正離子
具有芳香性或者α-位含有羥基、氨基的碳正離子是特別穩定的碳正離子,如片吶醇重排的驅動力是叔碳正離子重排為α-羥基碳正離子,表明α-羥基碳正離子比叔碳正離子還穩定。為了定量衡量這類碳正離子的穩定性,本文計算了如下7個碳正離子,它們分子中的共價鍵類型和數量都相同,所以能量差別主要來自于碳正離子的穩定性差別,結果見圖7。
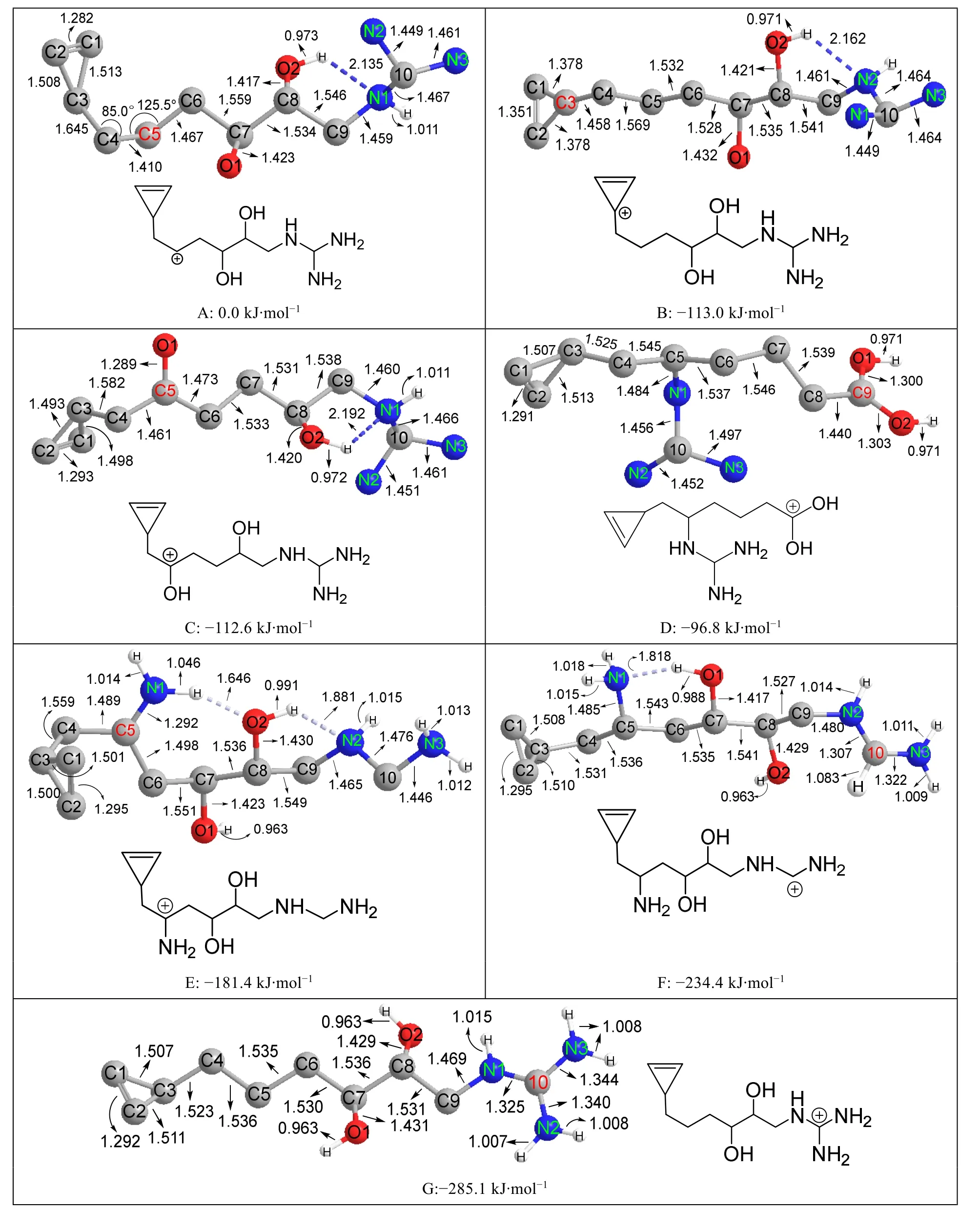
圖7 互為同分異構體的7個碳正離子的結構和能量
從圖7可以得到如下結果:
1) 以仲碳正離子的能量為參照(設為0.0 kJ·mol-1),可以看出其他6個碳正離子都比仲碳正離子穩定得多,它們與仲碳正離子的能量差值(約100-285 kJ·mol-1)都明顯超過叔碳正離子與仲碳正離子的能量差值(約50 kJ·mol-1),說明這些碳正離子都比叔碳正離子穩定得多。
2) 芳香正離子(本文為環丙基碳正離子)與單羥基碳正離子的穩定性相當。氨基碳正離子比單羥基碳正離子穩定得多,表明氨基對碳正離子的穩定作用明顯超過羥基。每增加一個氨基,化合物的能量降低約50 kJ·mol-1,所以脒鹽和胍鹽碳正離子都非常穩定,與實驗上發現胍和脒都是強堿的事實吻合。但是雙羥碳正離子(圖7D)反而不如單羥碳正離子(圖7C)穩定,原因可能是單羥碳正離子中有一個氫鍵作用(N1…HO2)而雙羥碳正離子沒有(圖7D中沒有氫鍵作用)。為了排除氫鍵數目不等造成的干擾,本文又計算了互為同分異構體且都沒有氫鍵作用的兩個碳正離子的能量,結果列于圖8。圖8中雙羥碳正離子(圖8B)比單羥碳正離子(圖8A)穩定約7.4 kJ·mol-1,這個計算結果與羧酸的堿性(質子化乙酸pKa= -6.1)強于醛酮(質子化丙酮pKa= -7.3)的實驗事實吻合。根據上述pKa差值估算的(ΔG= -2.303RTlgKa1/Ka2,按照Ka1/Ka2= 101.2、T= 300 K估算)質子化乙酸(具有雙羥碳正離子結構)和質子化丙酮(具有單羥碳正離子結構)穩定性差別約:2.303 × 2.493 × 1.2 = 6.9 kJ·mol-1,這個值與本文計算值(7.4 kJ·mol-1)非常接近,表明B3LYP方法在計算化合物能量方面比較可靠。由于雙羥基碳正離子只比單羥基正離子穩定7.4 kJ·mol-1,所以一個氫鍵的差別(氫鍵鍵能約20-25 kJ·mol-1)就足以使7C反而比7D穩定15.8 kJ·mol-1。

圖8 羥基碳正離子的結構和相對能量
3) 當NH2與OH位置靠近時,既可以形成O—H…N氫鍵,也能形成N—H…O氫鍵,從圖7A、7B、7C、7F看,形成的都是前者而不是后者,表明前者是比后者更強的氫鍵。從圖7F和圖7G來看,脒鹽或胍鹽中的氨基因為其孤對電子與碳正離子有強烈共軛效應,N原子的電子密度大大降低,已經不能再與鄰近的OH形成氫鍵,表明氫鍵的形成對受體的電子密度有較高要求。
4) OH和NH2在形成氫鍵時,O—H和N—H鍵長都會拉長,前者大約從0.0963 nm拉長到0.0988 nm,后者大約從0.1012 nm拉長到0.1046 nm。但是與碳正離子形成共軛效應后(即與碳正離子緊相鄰),O—H和N—H的鍵長變化方向不一樣。普通OH中O—H鍵長約0.0963 nm,與碳正離子共軛后拉長到0.0971 nm,如圖7D所示。普通NH2中N—H鍵長約0.1012 nm,與碳正離子共軛后縮短到0.1008 nm,如圖7G所示。導致這種現象的原因可能是OH與碳正離子共軛后呈現質子化酮的結構,質子化酮酸性很強,容易電離出H+,故O—H變長;但是NH2與碳正離子共軛后呈現質子化亞胺的結構,質子化亞胺酸性不強,不易電離出H+,NH2只好通過調整雜化狀態來加強共軛效應,從普通NH2的sp3雜化調整為sp2雜化,以便孤對電子所在的未雜化p軌道可以與碳正離子的未雜化空p軌道能更好重疊。由于sp2軌道的s成分比sp3高,更加收縮,故N—H鍵長較短,如同烯烴的C—H鍵長(通常約0.1085 nm)比烷烴的C—H鍵長(通常約0.1095 nm)略短一樣,如Scheme 4所示。對于雙羥碳正離子,第一個羥基氫原子的電離對第二個羥基氫原子的電離有抑制作用,所以第二個羥基對相鄰碳正離子的穩定作用不如第一個羥基大,導致雙羥碳正離子僅比單羥碳正離子穩定7.4 kJ·mol-1,不像脒鹽正離子與氨基碳正離子的穩定性差別能達到53.0 kJ·mol-1。
5) 圖7A中C3—C4的鍵長是0.1645 nm,圖7B中C3—C4的鍵長只有0.1458 nm,分別是圖7所有化合物中最長和最短的C—C單鍵(普通的C—C單鍵鍵長為0.1540 nm)。這種結構現象可以通過碳正離子的共振得到解釋,如Scheme 5所示。其中左圖仲碳正離子的共振結構還能解釋圖7A中∠C3—C4—C5只有85.0°的反常現象。

Scheme 5 圖7A (左)和圖7B (右)所示碳正離子的共振結構圖
4 結語
伯碳正離子比仲碳正離子不穩定得多,能量差值約60 kJ·mol-1;仲碳正離子又比叔碳正離子不穩定得多,能量差值約50 kJ·mol-1。普通的伯碳正離子在優化時總是重排為仲碳正離子,但乙基碳正離子是例外,因為它無法重排為仲碳正離子,所以采用超共價結構來避免形成伯碳正離子。仲碳正離子相對穩定,當它們不能重排為叔碳正離子時,它們也不形成超共價結構,而是形成強的超共軛效應。烯丙基型碳正離子都比較穩定,優化時不發生重排。其中仲和叔烯丙型碳正離子比叔碳正離子穩定。α-位有羥基或者氨基的碳正離子特別穩定,α-羥基碳正離子的穩定性與具有芳香性的環丙烯碳正離子相當,比叔碳正離子穩定約50 kJ·mol-1。α-氨基碳正離子比α-羥基碳正離子更穩定,能量差值約70 kJ·mol-1。在α-位每增加一個氨基,碳正離子能量約降低50 kJ·mol-1。但是α-位增加一個羥基對碳正離子能量的降低作用很有限,只有約7.4 kJ·mol-1。氨基和羥基的這個差別可能源自它們對碳正離子的不同的穩定作用機制。
猜你喜歡
房地產導刊(2022年5期)2022-06-01 06:20:14
哲學評論(2021年2期)2021-08-22 01:53:34
建材發展導向(2021年12期)2021-07-22 08:06:48
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中學生數理化(高中版.高考數學)(2021年12期)2021-03-08 01:28:50
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現代企業(2015年9期)2015-02-28 18:56:50
