SOD1基因p.H44R罕見位點突變致肌萎縮側(cè)索硬化癥1例報告并文獻(xiàn)復(fù)習(xí)
2023-12-18 04:55:30王雅歡劉洪雨何金婷王姣琦
中風(fēng)與神經(jīng)疾病雜志 2023年11期
關(guān)鍵詞:基因突變
王雅歡, 楊 偲, 劉洪雨, 何金婷, 王姣琦
運動神經(jīng)元病(motor neuron disease,MND)是一組起病隱匿的慢性進(jìn)行性神經(jīng)系統(tǒng)變性疾病,病因尚不十分清楚,可能與遺傳或環(huán)境因素相關(guān)。該病主要侵犯大腦皮質(zhì)錐體細(xì)胞、腦干運動神經(jīng)核及脊髓前角運動神經(jīng)元。肌萎縮側(cè)索硬化癥(amyotrophic lateral sclerosis,ALS)是最常見的一種,根據(jù)是否有家族史分為家族性肌萎縮硬化癥(familial amyotrophic lateral sclerosis,fALS)和散發(fā)性肌萎縮硬化癥(sporadic amyotrophic lateral sclerosis,sALS)。該病的早期表現(xiàn)是進(jìn)行性肌萎縮、肌無力和延髓麻痹,最終常累及呼吸肌導(dǎo)致呼吸衰竭死亡。5%~10%的患者有家族史,但大多數(shù)患者的病因仍不清楚[1]。該病不可逆轉(zhuǎn),且缺乏有效治療手段,其診斷為排除性診斷,需與表現(xiàn)相似的其他運動神經(jīng)元病相鑒別[2],早期診斷困難。現(xiàn)將吉林大學(xué)中日聯(lián)誼醫(yī)院神經(jīng)內(nèi)科收治的1 例以右下肢疼痛、無力為首發(fā)癥狀,通過肌電圖及全外顯子測序診斷的SOD1罕見位點突變致ALS 病例進(jìn)行回顧分析及文獻(xiàn)復(fù)習(xí),希望對該病在臨床上的早期診斷及預(yù)后評估有所幫助。
1 病例資料
患者,男,40 歲,因右下肢疼痛5 個月,無力4 個月,于2022年2月入院,5 個月前接種疫苗后出現(xiàn)右下肢肌肉酸痛,因未影響肢體活動而未在意,4 個月前出現(xiàn)右下肢無力、肉跳,逐漸發(fā)展并出現(xiàn)臀部、大腿及小腿肌肉萎縮,服用B 族維生素類藥物后癥狀未見緩解,近期使用筋膜槍理療,下肢無力的癥狀仍逐漸加重,入院前20 d出現(xiàn)右腿上臺階費力,其余肢體無明顯異常。病程中患者無頭暈、無智能減退、無動作遲緩、無腰部疼痛、無舌肌萎縮、無吞咽及呼吸困難,體重未見明顯變化。既往無基礎(chǔ)疾病。家族史:患者堂哥及2 個姑姑被診斷為“運動神經(jīng)元病”,但均已過世,無法提供臨床資料及相關(guān)檢查結(jié)果。患者父親因肺癌于48歲去世(患者家系圖見圖1)。

圖1 患者家系圖
入院后神經(jīng)系統(tǒng)查體:意識清楚,顱神經(jīng)查體未見明顯異常,右下肢屈髖肌力2級,伸髖肌力3級,屈膝肌力3 級,伸膝肌力4 級,遠(yuǎn)端背屈肌力2 級,跖屈肌力4 級,余肢體肌力未見異常,右下肢肌張力減弱,肌容積減少,右下肢膝腱反射減弱,跟腱反射未引出,無感覺異常,雙側(cè)病理反射陰性,余查體未見異常。
實驗室檢查:肌酸激酶600 U/L(正常值<164 U/L),肌酸激酶同工酶17.00 ng/ml(正常值2.00~7.20 ng/ml),肌紅蛋白149 ng/ml(正常值23.00~112.00 ng/ml),D-二聚體3 340.00 ng/ml(正常值80~500 ng/ml)。甲功3 項、降鈣素原、C 反應(yīng)蛋白、常規(guī)免疫、腫瘤標(biāo)志物、免疫球蛋白+補(bǔ)體、風(fēng)濕免疫相關(guān)抗體未見明顯異常。頭部核磁未見異常。腰骶叢神經(jīng)MR 平掃+增強(qiáng):左側(cè)骶1、2 神經(jīng)根束膜囊腫可能性大,腰椎MR:腰5-骶1 椎間盤輕度突出。肺部CT:左肺下葉外基底段結(jié)節(jié),性質(zhì)傾向良性。腹部超聲:脂肪肝。肌電圖示:右下肢呈神經(jīng)源性改變(根或以上水平受損可能);左脛前肌、左腓腸肌可見神經(jīng)源性改變(見圖2)。進(jìn)一步完善的腰穿腦脊液檢查示腦脊液無色透明,細(xì)胞總數(shù)4×106/L,白細(xì)胞數(shù)4×106/L,血抗神經(jīng)節(jié)苷脂抗體陰性。提檢全外顯子測序,結(jié)果回報前初步診斷為下運動神經(jīng)元綜合征,待除外肌萎縮側(cè)索硬化癥。給予患者營養(yǎng)神經(jīng)、針灸、理療、被動康復(fù)訓(xùn)練等對癥治療,患者自覺右踇趾背屈稍有改善后出院。
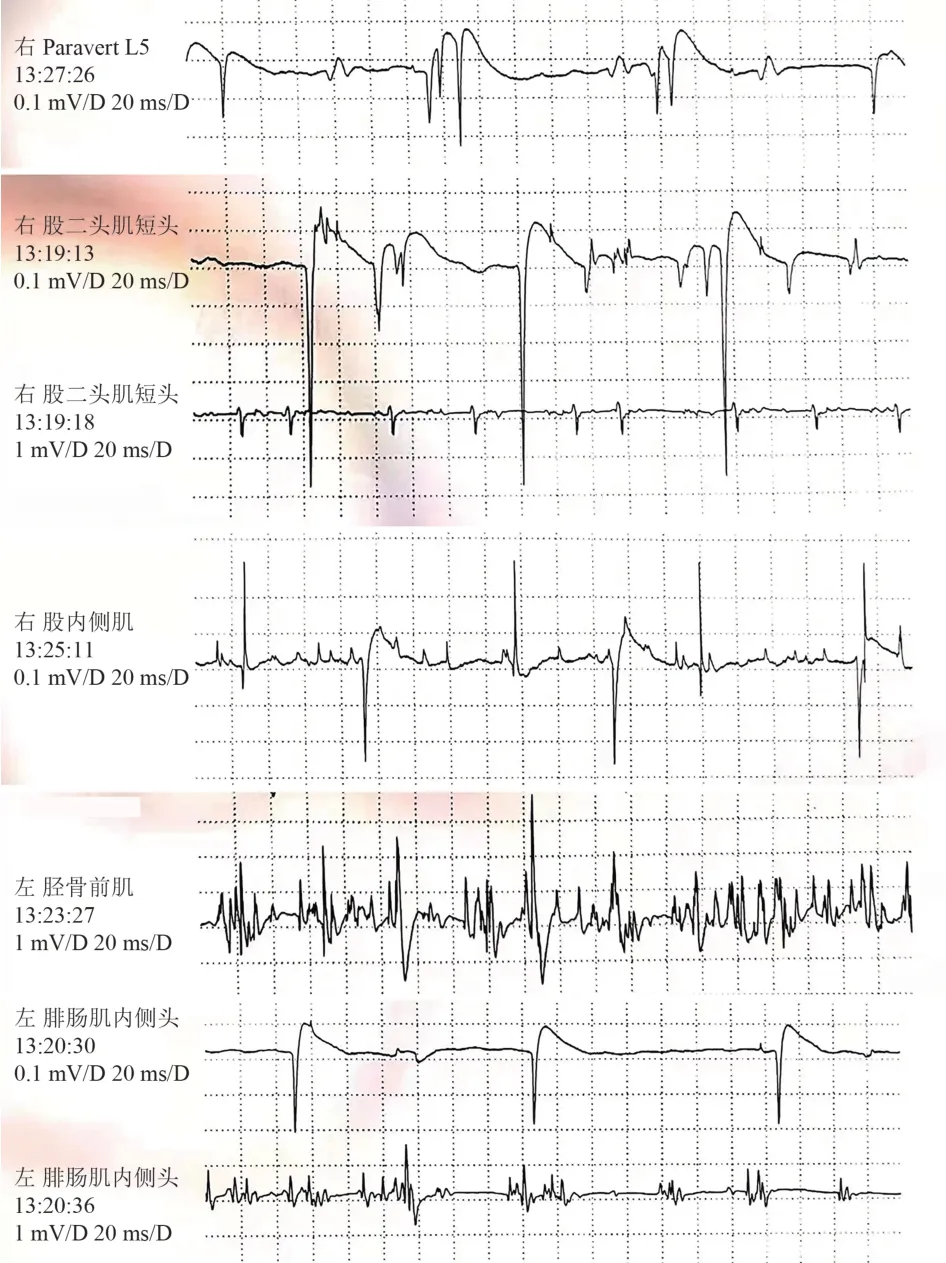
圖2 患者的肌電圖結(jié)果
全外顯子測序提示患者攜帶SOD1基因上的一個雜合錯義變異:c. 131A>G:p. H44R。由于先證者父母未能配合驗證,變異來源未知。該變異為cDNA的第131 位堿基由A 替換為G,導(dǎo)致SOD1基因第44位密碼子由編碼組氨酸變?yōu)榫幋a精氨酸(見圖3)。
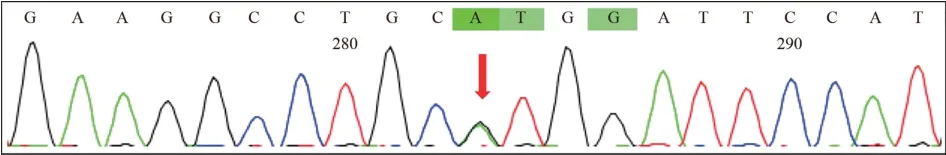
圖3 患者的一代測序峰圖
獲得全外顯子測序結(jié)果后,從下運動神經(jīng)元綜合征入手剖析患者的診斷。患者無中毒、輻射,無脊髓灰質(zhì)炎、艾滋病、腸道及水痘帶狀皰疹病毒感染史,無蜱叮咬史,腦脊液細(xì)胞數(shù)正常,可除外中毒、感染、輻射等病因,重點篩查免疫、變性及遺傳方面。患者偏側(cè)受累,抗神經(jīng)節(jié)苷脂抗體陰性,不符合吉蘭-巴雷綜合征(guillain-barré syndrome, GBS);患者腫瘤標(biāo)志物陰性,腹部超聲、胸部CT 篩查未見占位性病變,暫不支持副腫瘤性下運動神經(jīng)元綜合征;患者肌電圖無局灶性運動傳導(dǎo)阻滯,且早期出現(xiàn)的明顯的肌肉萎縮也不支持多灶性運動神經(jīng)病(multifocal motor neuropathy,MMN),故可除外免疫因素。變性方面,單肢肌萎縮(monomelic muscular atrophy,MMA)為自限性,多見于青少年,累及上肢時又稱平山病,該病很少累及對側(cè)肢體,與患者的發(fā)病年齡及肌電圖上左下肢亞臨床神經(jīng)源性損害不符。遺傳方面,遺傳性遠(yuǎn)端運動神經(jīng)病(distal hereditary motor neuropathy,dHMN)及脊髓性肌萎縮(spinal muscular atrophy,SMA)為單基因病,全外顯子測序結(jié)果不支持。最后,從遺傳變性的角度需要鑒別ALS 和進(jìn)行性肌萎縮(progressive muscular atrophy,PMA)。PMA主要表現(xiàn)為進(jìn)行性肌無力或萎縮,不伴上運動神經(jīng)元功能障礙,該病比其他類型運動神經(jīng)元病的進(jìn)展慢,暫不能完全除外。患者全外顯子測序提示攜帶SOD1基因上的一個雜合錯義變異。目前國內(nèi)尚未將基因檢測納入ALS 診斷標(biāo)準(zhǔn),但是查閱文獻(xiàn)發(fā)現(xiàn)該突變是ALS 已知的致病突變,呈常染色體顯性遺傳。2015年修訂的E1 Escorial 診斷標(biāo)準(zhǔn)中提出,若基因檢測發(fā)現(xiàn)患者攜帶已知的肌萎縮側(cè)索硬化癥致病基因,且具有一個區(qū)域的上運動神經(jīng)元或下運動神經(jīng)元損害證據(jù)可診斷為ALS,如果至少有一個一級或二級親屬患有ALS 則考慮為遺傳性ALS[3]。患者查體見下運動神經(jīng)元損傷證據(jù),并有基因檢測支持,故排除PMA,診斷為ALS。囑患者服用利魯唑,隨訪。后患者就診于北京大學(xué)第三醫(yī)院,維持ALS診斷。2 個月后電話隨訪,患者出現(xiàn)了左下肢的無力。
2 討 論
ALS 常中年以后隱蔽起病,進(jìn)展緩慢,是一種上、下運動神經(jīng)元同時受累的神經(jīng)系統(tǒng)變性疾病,通常以肌無力、萎縮等為主要表現(xiàn),受累部位常有肌束震顫,伴有腱反射亢進(jìn)、病理反射陽性。大多數(shù)患者最終因呼吸衰竭死亡。目前國際公認(rèn)的ALS診斷標(biāo)準(zhǔn)共有3 種,依次為E1 Escorial 診斷標(biāo)準(zhǔn)、Airlie House 診斷標(biāo)準(zhǔn)和Awaji-shima 電生理診斷標(biāo)準(zhǔn),目前臨床上應(yīng)用最廣的是Airlie House 診斷標(biāo)準(zhǔn),該標(biāo)準(zhǔn)將ALS 診斷等級分為確診級肌萎縮側(cè)索硬化、擬診級肌萎縮側(cè)索硬化、實驗室支持?jǐn)M診級肌萎縮側(cè)索硬化、可能級肌萎縮側(cè)索硬化[4]。基于以上診斷標(biāo)準(zhǔn),極大地提高了ALS 患者的診斷效率和病情評判。根據(jù)患者的臨床表現(xiàn),最新的ALS 分型將肌萎縮側(cè)索硬化分為8型[5]:(1)經(jīng)典型,以上肢或下肢肌無力為首發(fā)癥狀,錐體束征常不明顯,是男性患者中最常見的類型,平均發(fā)病年齡在62.8 歲,10年生存率為13%,伴發(fā)額顳葉癡呆(frontotemporaldementia,F(xiàn)TD)的占4%;(2)延髓型,以構(gòu)音障礙和(或)吞咽困難等延髓損害為特點,發(fā)病6 個月內(nèi)錐體束征可不明顯,隨著疾病發(fā)展,錐體束征可越來越明顯,男性發(fā)病率略低于女性,10年生存率僅為3.4%,F(xiàn)TD發(fā)生率最高為9%;(3)連枷臂型,以上肢近端肌無力和萎縮起病,可伴有上肢的腱反射活躍及Hoffman征,肌張力正常,發(fā)病后,功能受累必須局限于連枷肢體至少12個月,男女發(fā)病比例為4∶1,10年存活率為17.4%,很少伴發(fā)FTD,僅占1.4%;(4)連枷腿型,主要為雙下肢進(jìn)行性肌無力和萎縮,病程中下肢可有腱反射活躍或Babinski 征,肌張力正常。注意除外無遠(yuǎn)端受累的患者,這種情況常提示為經(jīng)典型。男女發(fā)病比例為1.03∶1,10年存活率為12.8%,合并FTD 的占4.1%;(5)錐體束型,以錐體束征為主,如嚴(yán)重的痙攣性截癱或四肢癱,可出現(xiàn)在疾病早期或晚期,常累及至少兩個不同區(qū)域,出現(xiàn)明顯的下運動神經(jīng)元損害的體征,如肌肉無力和萎縮。肌電圖檢查存在慢性和活動性的失神經(jīng)損害。此種表型發(fā)病較早,常于58.3 歲左右,男女比例為1.04∶1,F(xiàn)TD較少見,占2.5%,10年存活率為31.9%;(6)呼吸型,此類型患者普遍存在呼吸障礙,表現(xiàn)為呼吸困難或端坐呼吸,發(fā)病半年內(nèi)脊髓或球部受累癥狀較輕,可伴有上運動神經(jīng)元損害跡象。該型為最罕見的表型,男性發(fā)病率0.06/10 萬,女性發(fā)病率0.01/10 萬,無1例患者存活10年以上;(7)純下運動神經(jīng)元綜合征(pure lower motor neuron syndrome,PLMN),病變累及腦神經(jīng)運動核和脊髓前角細(xì)胞,典型表現(xiàn)為肌無力或萎縮、腱反射減弱,但感覺不受累,男性發(fā)病率是女性2 倍,不伴有FTD,預(yù)后與其他表型相比較好,10年生存率為36.6%;(8)純上運動神經(jīng)元綜合征(pure upper motor neuron syndrome,PUMN),臨床表現(xiàn)包括痙攣性截癱和(或)四肢癱,病理反射亢進(jìn),腱反射活躍,言語障礙等,此種表型發(fā)病率較低(男女均為0.12/10 萬),存活時間最長(13.1年),10年生存率為71.1%。此外還有學(xué)者認(rèn)為原發(fā)側(cè)索硬化(primary lateral sclerosis,PLS)和進(jìn)行性肌萎縮(progressive muscular atrophy,PMA)為ALS 的特殊類型,PLS 較為少見,一般中年起病,4年內(nèi)僅有上運動神經(jīng)元受累表現(xiàn);PMA僅有下運動神經(jīng)元受累表現(xiàn),以男性患者多見,發(fā)病年齡晚,平均生存期比ALS顯著延長。以上兩種類型隨著疾病的發(fā)展會出現(xiàn)上、下運動神經(jīng)元同時受累的情況,與ALS表現(xiàn)相似,所以上述兩種類型可以被歸為ALS 的特殊類型[4]。ALS復(fù)雜的臨床分型不僅說明了該病在起病形式、受累模式、流行病學(xué)等方面的多樣性,而且也提示了該病病因的復(fù)雜性。
目前發(fā)現(xiàn)與fALS發(fā)病相關(guān)的基因約30多種,包括銅/鋅超氧化物歧化酶(SOD1)基因、反式激活反應(yīng)-DNA 結(jié)合蛋白(TARDBP)基因、肉瘤熔合(FUS)基因等[6]。其中,SOD1基因突變是我國sALS 和fALS 最常見的突變類型,在sALS 患者中占1%~2%,fALS 占25%[7]。1993年Rosen 等首次發(fā)現(xiàn)SOD1突變引發(fā)ALS[8]。SOD1是一種由153 個氨基酸組成的酶,在神經(jīng)系統(tǒng)、肝臟及紅細(xì)胞中高表達(dá),參與自由基清除。目前已知的與ALS 相關(guān)的SOD1基因突變超過150個[9]。本文列舉了常見的幾種SOD1基因突變類型、遺傳方式及臨床表現(xiàn)(見表1)。不同突變型引發(fā)的ALS在起病時間、受累部位、病程進(jìn)展速度等方面都存在差異。其中A4V突變是最常見的SOD1基因突變,常表現(xiàn)為下運動神經(jīng)元損傷,平均存活時間僅1.5年。D90A突變提示上運動神經(jīng)元損傷較下運動神經(jīng)元損傷明顯,且疾病進(jìn)展較慢,患者存活時間相對較長。G37R、G41D、G93C突變提示患者存活期較長,G37R、L38V、L106V突變提示患者發(fā)病年齡較小。其中,L106V基因突變患者的發(fā)病年齡是目前已知最年輕的,為35.5 歲,而I113T基因突變的發(fā)病年齡最晚,為58.9 歲[10]。大多數(shù)ALS 患者在癥狀出現(xiàn)后3~5年內(nèi)死亡,但變異性很大,有些患者在發(fā)病后幾個月死亡,而有些患者甚至存活了20年。即使是來自于一個家族的不同個體,雖然具有完全相同的基因突變位點,但其生存期和發(fā)病年齡上也有很大的差異,這說明還存在其他改變表型的因素有待進(jìn)一步發(fā)現(xiàn)[9]。

表1 常見SOD1基因突變位點及其遺傳方式、臨床表現(xiàn)
本例患者以右下肢疼痛、無力伴肌肉萎縮起病,感覺系統(tǒng)無陽性體征,癥狀暫未累及其他肢體,且無球部損傷癥狀,定位在脊髓前角及前根。肌電圖示右下肢神經(jīng)源性改變(根或以上水平),左側(cè)脛前肌、腓腸肌可見自發(fā)電。該肌電圖結(jié)果一方面支持上述定位;另一方面也提示左下肢出現(xiàn)亞臨床的急性神經(jīng)源性損害。患者腰骶叢核磁、腰椎核磁未見明確的脊髓及神經(jīng)根病變,故除外神經(jīng)根損傷,定位在脊髓前角細(xì)胞。患者病前有疫苗接種史,有可疑ALS家族史,定性上應(yīng)重點考慮免疫因素及遺傳變性病。根據(jù)下運動神經(jīng)元綜合征的病因分析,患者免疫相關(guān)等實驗室檢查結(jié)果未見異常,影像學(xué)及腦脊液檢查未見炎性改變,暫不支持免疫炎癥,故遺傳變性病的可能性大,不除外以下運動神經(jīng)元損傷起病的ALS。患者存在ALS 家族史,雖然無法明確證實,但從一元論的角度考慮,基因測序有望為患者早期診斷提供依據(jù)。當(dāng)患者出現(xiàn)下運動神經(jīng)元綜合征的表現(xiàn)并疑診ALS 時,完善非受累肢體或節(jié)段的肌電圖檢查可提供臨床前的下運動神經(jīng)元損傷證據(jù),為了解疾病全貌,對疾病進(jìn)行早期診斷提供幫助。全外顯子測序提示患者SOD1基因突變,最終診斷為ALS。該患者為雜合子突變,文獻(xiàn)報道ALS突變類型大多數(shù)是常染色體顯性遺傳,D90A、D96N基因突變是常染色體隱性遺傳或常染色體顯性遺傳[23],基因檢測結(jié)果提示患者為SOD1基因第二外顯子c. 131A>G:p.H44R突變,該變異為罕見變異。關(guān)于H44R基因突變致ALS的報道最早可以追溯到1995年[24]。2003年首次對該突變致ALS 的臨床表現(xiàn)進(jìn)行了詳細(xì)的描述,該患者是名58 歲的日本女性,因下肢單癱就診,有ALS家族史,發(fā)病7個月后死于呼吸衰竭。該患者家族系譜顯示為常染色體顯性遺傳,具有完全外顯率[25]。2017年,該基因突變位點在我國患者中首次報道,1 例47 歲男性以上肢無力起病,9 個月后因呼吸衰竭死亡,不同的是此患者無家族史,該變異為SOD1基因上的一個雜合錯義突變[7]。本例患者的發(fā)病年齡較前2 例更早,單肢癱起病,臨床癥狀雖然局限,但肌電圖已提示其他肢體的早期受累。目前患者病程較短,還有待進(jìn)一步隨訪以明確患者病情進(jìn)展及預(yù)后情況。結(jié)合已報道的病例特點,推測H44R基因突變導(dǎo)致的ALS 可能主要以下運動神經(jīng)元損傷為主,可有球部受累癥狀,病情進(jìn)展快,生存期可能較短。
盡管目前美國FDA已批準(zhǔn)利魯唑作為ALS的首選治療藥物,但其療效一般,僅延長2~3 個月的無氣管切開術(shù)生存期[26],患者最終走向死亡。2022年我國ALS診斷和治療專家共識提示基因檢測陽性可加速ALS 的診斷進(jìn)程,使患者盡早開始接受藥物治療[27]。隨著測序技術(shù)的發(fā)展,基因檢測的經(jīng)濟(jì)成本越來越低,該技術(shù)對廣大民眾的可及性也在提高。在使用該技術(shù)早期診斷治療的同時,我們也要警惕技術(shù)濫用及隨之而來的倫理問題。
倫理學(xué)聲明:本研究經(jīng)由吉林大學(xué)中日聯(lián)誼醫(yī)院倫理委員會審批(審批號:2023111001)。
利益沖突聲明:所有作者均聲明不存在利益沖突。
作者貢獻(xiàn)聲明:王雅歡負(fù)責(zé)采集數(shù)據(jù)、數(shù)據(jù)整理及分析、論文撰寫;楊偲、劉洪雨、何金婷負(fù)責(zé)采集數(shù)據(jù)、分析數(shù)據(jù)、研究指導(dǎo);王姣琦負(fù)責(zé)研究指導(dǎo)、論文修改。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫(yī)學(xué)影像學(xué)雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學(xué)生導(dǎo)刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現(xiàn)代檢驗醫(yī)學(xué)雜志(2016年4期)2016-11-15 02:01:14
中國現(xiàn)代醫(yī)學(xué)雜志(2015年26期)2015-12-23 11:04:22
鄭州大學(xué)學(xué)報(醫(yī)學(xué)版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經(jīng)精神疾病雜志(2014年1期)2014-03-01 03:23:22
