SFOD-LPME-HPLC法測定白酒中甘草酸、甘草次酸
2023-09-28 03:50:00黃何何胡朝陽蔡小明
中國釀造 2023年9期
黃 媛,洪 麗,黃何何,潘 城,胡朝陽,蔡小明*
(福建省產品質量檢驗研究院 國家加工食品質量監督檢驗中心,福建 福州 350001)
白酒被譽為世界六大蒸餾酒之一[1],源自我國傳統的釀造工藝,具有醇香甜美的獨特口感。而現代生產工藝追求效益,大多發酵時間短,在白酒制作過程中產生多元醇或酮等天然甜味物質一般都達不到理想的效果,部分不良商家為了以次充好,在白酒中添加甜味劑來掩蓋苦味[2]。我國標準GB 2760—2014《食品添加劑使用標準》中明確規定,白酒中不得添加任何甜味劑[3]。關于白酒中檢出甜蜜素、糖精鈉、安賽蜜等常見甜味劑的報道屢見不鮮[4-6],隨著近幾年監管的加強,對于白酒中違規添加人工甜味劑的現象已經越來越受到重視,為了逃避監管,商家極可能選擇新型的不在監測范圍內的甜味劑進行使用[7]。因此對于能夠應用于白酒中的天然甜味劑也應當引起相關部門的關注與重視。
甘草酸(glycyrrhizin,GA)、甘草次酸(glycyrrhetinic acid,GTA)屬于三萜類化合物,甜度約為蔗糖的80~300倍[8]。因其高甜、低熱、安全的特點,在食品工業中作為甜味劑得到了廣泛的應用[9-10]。鑒于其優越的甜度,在食品中添加少量即可達到口感需求。目前,食品中甘草酸、甘草次酸檢測參考依據是SN/T 3854—2014《出口食品中天然甜味劑甜菊糖苷、甜菊雙糖苷、甘草酸、甘草次酸的測定》[11],該方法前處理繁雜,且檢出限高。報道顯示,白酒中檢出的甜味劑往往是少量,甚至微量,使用現行標準方法難以滿足行業的檢測需求。目前關于甘草酸、甘草次酸的研究大多在其藥理學及臨床學等領域,有關含量檢測的相關報道較少。而白酒中甜味劑檢測方法的研究大多集中于高效液相色譜-質譜聯用法(high performance liquid chromatography-mass spectrometry,GC-MS)[12-14],該方法能夠實現多種甜味劑的聯測,可達到較低的檢出限,但天然甜味劑的檢出限也通常高于人工甜味劑[1],與市場實際添加情況相悖。液質聯用法具有強大的定性功能,但由于樣品前處理多采用樣液直接或稀釋后上機,不可避免產生基質效應,因此在定量上不如高效液相色譜準確,同時,液質聯用法對操作人員要求較高,不利于方法的推廣。因此,開發一種簡單、快速、準確的方法測定白酒中天然甜味劑成為目前重要的一個研究方向。
漂浮有機液滴凝固液相微萃取(liquid phase microextraction based on solidification of floating organic drop,SFODLPME)是近幾年液相微萃取技術中發展的比較好的一個分支[15-16],該方法采用小體積有機試劑作為萃取劑,對萃取供相中的分析物進行富集,而后利用萃取劑密度比水小且溶點接近室溫的特性達到相分離。方法綠色環保,方便快捷,具有卓越的富集能力,特別適用于痕量物質的提取分離,目前已在食品安全檢測領域得到應用研究[17-20]。本研究將SFOD-LPME應用于白酒中甘草酸、甘草次酸的富集,并聯合高效液相色譜(HPLC)技術進行測定,為白酒中甜味劑檢測方法的建立及優化提供技術參考。
1 材料與方法
1.1 材料與試劑
甘草酸(純度98%):成都艾科達化學試劑有限公司;甘草次酸(純度97%):百靈威科技有限公司;正十一醇(純度>99%)、正十二醇(純度>98%)、1,10-二氯葵烷(純度≥99.5%):上海皓鴻生物醫藥科技有限公司;丙酮、四氫呋喃、甲醇、乙腈(均為色譜純)、乙醇、鹽酸(均為分析純):國藥集團化學試劑有限公司;除特殊說明外,實驗用水均為超純水;白酒樣品:隨機采購于各大商超及零售店。
1.2 儀器與設備
WatersARCHPLC超高效液相色譜儀(配備二極管陣列檢測器):美國Waters公司;RD-C18色譜柱(250 mm×4.6 mm,5 μm):中譜科技有限公司;HWS-26電熱恒溫水浴鍋:上海一恒科學儀器有限公司;HJ-6A多頭恒溫磁力攪拌器:國華(常州)儀器制造有限公司。
1.3 方法
1.3.1 標準溶液配制
分別準確稱取10 mg甘草酸、甘草次酸標準物質于容量瓶中,用甲醇稀釋定容至10 mL,得甘草酸、甘草次酸標準儲備液,于-18 ℃冷凍保存;根據需要將標準儲備液用水稀釋至一定濃度作為標準工作液,現用現配。
1.3.2 樣品處理
準確移取2 mL白酒置于50 mL玻璃比色管中,于95 ℃水浴10 min,加水溶液20 mL至比色管中進行復溶,渦旋混勻,作為待萃取樣品工作液(即萃取供相)。
SFOD-LPME條件:依次在樣品工作液加入100 μL 1∶1鹽酸溶液(供相溶液pH值約為3)、1.0 g氯化鈉、50 μL四氫呋喃以及40 μL正十二醇,將比色管置于多頭恒溫磁力攪拌器中,在40 ℃攪拌萃取15 min,后停止攪拌,待萃取劑在液面上匯聚成滴,將比色管轉移置于冰浴中,待正十二醇凝固后取出,室溫下融化,準確吸取30 μL萃取劑加入30 μL甲醇混勻后注入色譜系統。
另取20 mL標準工作液進行相同的SFOD-LPME萃取過程。
1.3.3 色譜條件
流動相:A相為0.02 mol/L磷酸水溶液,B相為乙腈;梯度洗脫程序見表1;流速:1 mL/min;進樣量:10 μL,柱溫:35 ℃,檢測波長:254 nm。
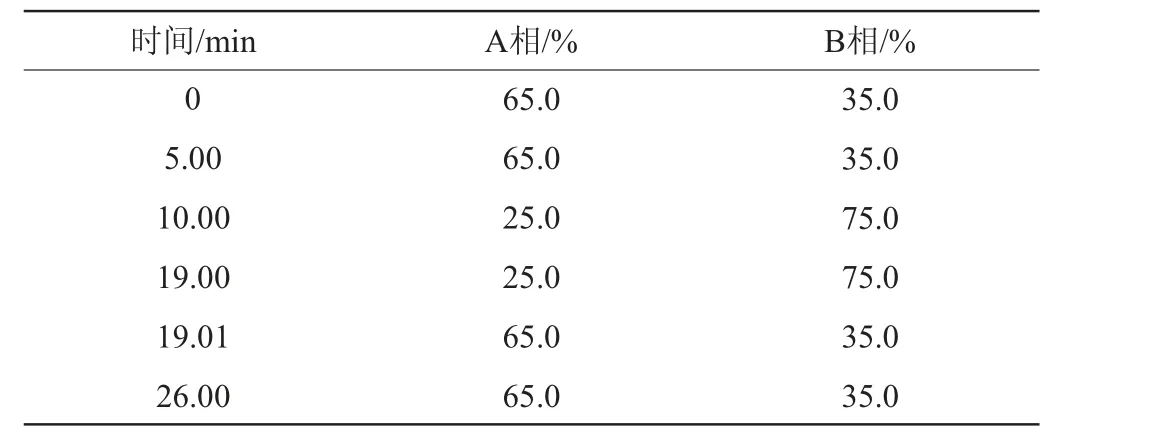
表1 液相色譜梯度洗脫程序Table 1 Gradient elution program of liquid chromatography
1.3.4 萃取條件優化
取20 mL水溶液作為萃取供相,采用單因素控制變量法,分別考察萃取劑(正十二醇、正十一醇以及1,10-二氯癸烷)、萃取劑體積(30~80 μL)、分散劑(四氫呋喃、乙腈、甲醇、丙酮)、分散劑體積(0~600 μL)、供相溶液中鹽酸添加量(0~250 μL)、氯化鈉添加量(0~2.0 g)、萃取溫度(26~60 ℃)、萃取時間(5~30 min)對萃取效率的影響,萃取完成后注入色譜系統進行測定,記錄峰面積,以峰面積大小作為萃取效率高低的判斷標準,用來確定最佳SFODLPME萃取條件。
1.3.5 方法學評價
(1)標準曲線回歸方程、檢出限及定量限
分別取適量甘草酸、甘草次酸標準儲備液,用水配制成系列質量濃度標準工作液,而后使用最佳的SFOD-LPME條件進行萃取,注入色譜系統進行分析,得到甘草酸、甘草次酸色譜圖及峰面積,以標準溶液質量濃度(x)為橫坐標,峰面積(y)為縱坐標,制作標準曲線;注入樣品萃取空白,計算信噪比,分別以3倍信噪比及10倍信噪比對應的被測物的濃度值,計算得該方法的檢出限及定量限。
(2)方法精密度及回收率考察
選取4種不同香型的白酒樣品作為加標基質,進行低、中、高3水平加標試驗,采用本方法萃取后進行測定,計算精密度試驗結果相對標準偏差(relative standard deviation,RSD)及平均回收率,進而完成該方法精密度及準確度考察。
1.3.6 數據分析
采用Empower色譜工作站進行定量分析,Microsoft Excel 2016 統計分析處理數據,Origin 9.5進行圖譜處理。
2 結果與分析
2.1 萃取條件優化
2.1.1 萃取劑的選擇
SFOD-LPME要求萃取劑密度小于水,并且溶點接近室溫,實驗考察了滿足條件的正十二醇、正十一醇以及1,10-二氯癸烷三種試劑對萃取效率的影響。萃取過程顯示,冰浴后,在萃取劑凝固狀態的完整性上,正十二醇優于正十一醇及1,10-二氯癸烷,冰浴凝固速度快,操作方便;而1,10-二氯癸烷萃取完成后,液體略呈白色渾濁狀態,萃取劑回收較差,可能由于其密度與水最為接近,導致部分滯留于樣品溶液中,與水相分離不完全;且色譜圖結果顯示,使用正十一醇萃取后,目標峰型展寬較大,疑存在萃取干擾。因此選擇最佳萃取劑為正十二醇。
在液相微萃取中,兩相間的傳質系數隨著樣品溶液體積和萃取劑體積的減少而增大[21],本研究在操作可行的條件下控制樣品溶液體積即萃取供相體積為20 mL,并在此體積下進行萃取劑體積的優化,取30~80 μL萃取劑進行萃取效率的考察,考察萃取劑對富集效果的影響。試驗結果顯示,萃取劑體積越大,相應富集倍數越小,當體積為30 μL時,萃取結果的平行性較差,萃取劑回收重復性低,當體積達到40 μL時,萃取劑回收情況開始呈現穩定狀態,綜合考慮,確定40 μL正十二醇作為萃取劑。
2.1.2 分散劑的選擇
分散劑一般選擇既溶于供相,又溶于萃取相的試劑,可以很好的將互不相溶的兩相進行充分的混合,在液相微萃取中起著至關重要的作用。該研究選取了四氫呋喃、乙腈、甲醇、丙酮四種溶劑為分散劑,以相同的添加量(100 μL)進行測試,并以不加分散劑為空白試驗,結果見圖1。選出最佳分散劑后,考察分散劑體積對萃取效果的影響,結果見圖2。
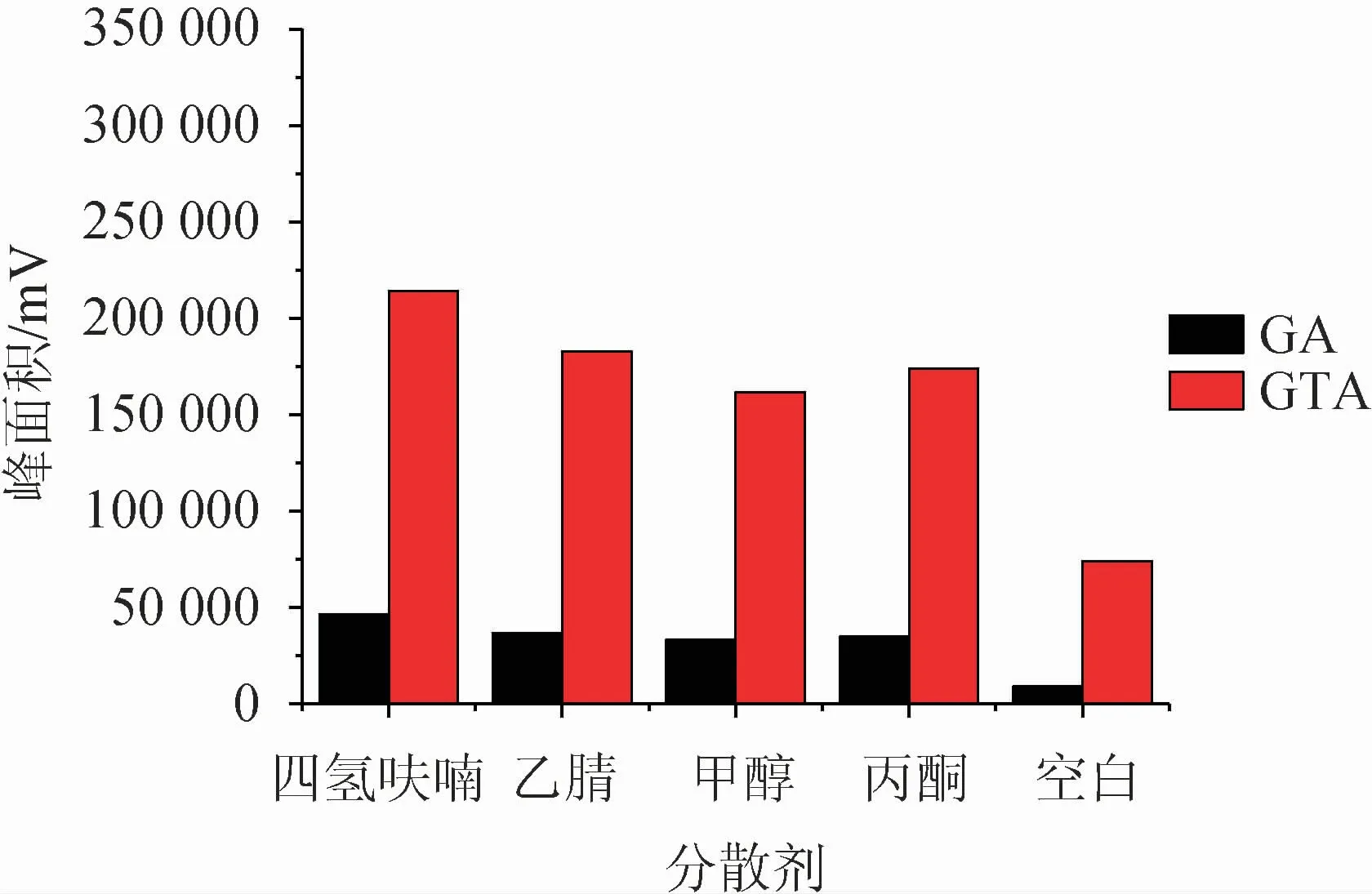
圖1 分散劑種類對萃取效果的影響Fig.1 Effect of dispersant varieties on extraction efficiency
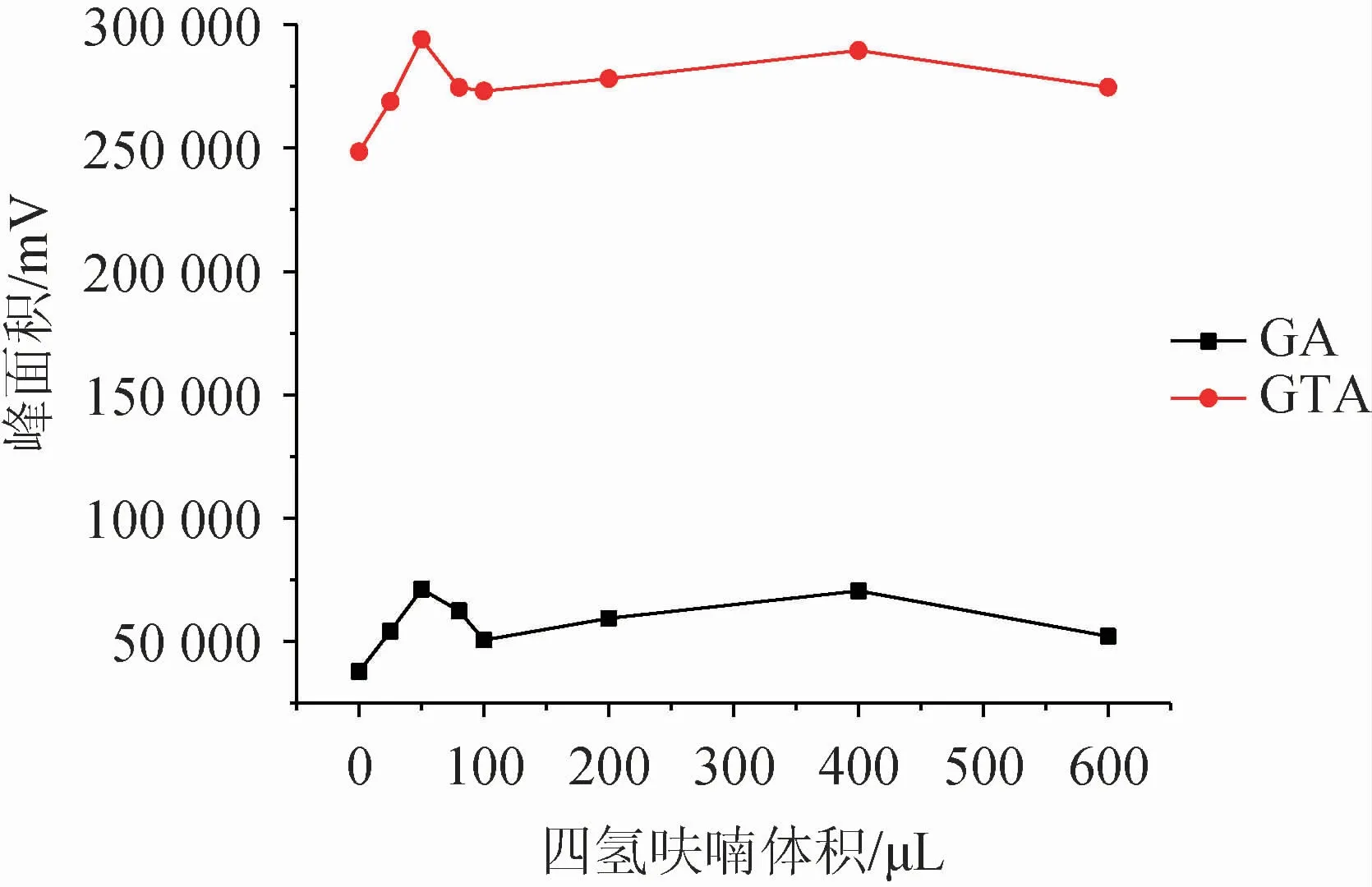
圖2 分散劑體積對萃取效果的影響Fig.2 Effect of dispersant volume on extraction efficiency
由圖1可知,在不加分散劑的情況下,目標物萃取效果明顯很差;而四種溶劑對甘草酸的萃取影響無明顯差異,對甘草次酸的影響較大,其中四氫呋喃對該方法體現出較好的適配性。因此選擇最佳分散劑為四氫呋喃。由圖2可知,隨著四氫呋喃添加量的增大,萃取效率逐漸增大,在分散劑體積為50 μL時,萃取效果最佳,隨之下降,繼續增加四氫呋喃添加量,萃取效果稍有增加但整體趨勢趨于平緩。綜合考慮,最終確定最佳分散劑為50 μL四氫呋喃。
2.1.3 鹽酸添加量的選擇
甘草酸由兩分子葡萄糖醛酸和一分子甘草次酸組成,和甘草次酸一樣,水溶液均呈弱酸性。因此,方法保持供相溶液為酸性體系,使目標化合物呈分子狀態,得以快速轉移進入萃取相中。該研究以20 mL水溶液為供相體系,考察了1∶1鹽酸水溶液的添加量(0~250 μL)對萃取效率的影響,結果見圖3。由圖3可知,不進行供相溶液酸度控制時,萃取后甘草酸幾乎無響應,說明甘草酸完全沒有被富集;隨著鹽酸添加量的增加,萃取效率逐漸增大,到一定程度時,則呈下降趨勢。當添加量為100 μL時,目標化合物富集效果最好,此后隨鹽酸添加量增大而降低,其中甘草次酸降低幅度較大。故確定鹽酸添加量為100 μL。
吉林省、四川省、浙江省等出臺的孤兒救助的社會政策也對孤兒教育做了較為明確的規定。通過梳理相關政策可以發現,對處于義務教育階段的孤兒,中央及各地方的教育保障政策都很明確,即實施免費義務教育。而對在普通高中、中等職業學校、高等職業學校和普通本科高校就讀的孤兒,除了北京提出免收學費、住宿費、服務性費用,天津提出免收學雜費外,中央和其他地方只是提出將這部分孤兒優先納入資助政策體系。對于成年孤兒在校就讀的,中央和地方文件都只有原則性的規定,即“孤兒成年后仍在校就讀的,繼續享有相應政策”。由此可見,對于大齡孤兒或者成年孤兒的教育救助或保障政策還需要進一步細化。

圖3 鹽酸添加量對萃取效果的影響Fig.3 Effect of hydrochloric acid addition on extraction efficiency
2.1.4 氯化鈉添加量的選擇
在供相溶液中加入一定量的鹽,可有效抑制目標化合物的離子化過程,從而增大富集效應。該研究比較了20 mL水溶液為供相體系下氯化鈉添加量為0~2.0 g情況下的萃取效果,結果見圖4。由圖4可知,隨著氯化鈉添加量的增加,萃取效率有一定改善,當添加量為1.0 g時,甘草酸和甘草次酸均得到了較好的響應,說明此時的鹽析效應最有利于萃取的進行,而隨著添加量的增大,萃取效率逐漸下降,可能是離子強度過強導致鹽離子在萃取劑中產生了靜電效應阻止了目標化合物進入萃取劑中[22]。故選擇氯化鈉添加量為1.0 g。
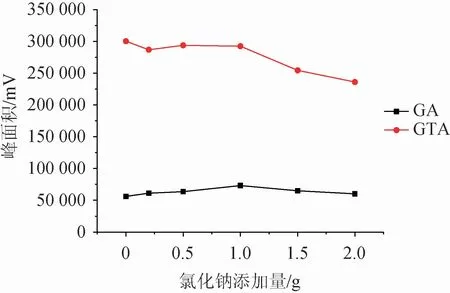
圖4 氯化鈉添加量對萃取效果的影響Fig.4 Effect of sodium chloride addition on extraction efficiency
2.1.5 萃取溫度的選擇
溫度提高能夠加快萃取體系中目標化合物的轉移,加速達到萃取平衡。實驗從室溫(26 ℃)開始,逐漸升高體系溫度,考察萃取溫度(26~60 ℃)對甘草酸、甘草次酸富集效率的影響,結果見圖5。由圖5可知,溫度對目標化合物萃取效率影響較大,響應值隨著溫度增高明顯呈上升趨勢。萃取溫度40 ℃時,目標化合物均得到最佳響應,而后下降,疑為溫度過高導致萃取劑揮發損失,回收率受影響,正十二醇萃取后液滴表面積隨溫度升高而增大,到達60 ℃時已呈平鋪狀態,不利于液滴的分離,實驗重復性呈下降趨勢。故確定萃取溫度為40 ℃。
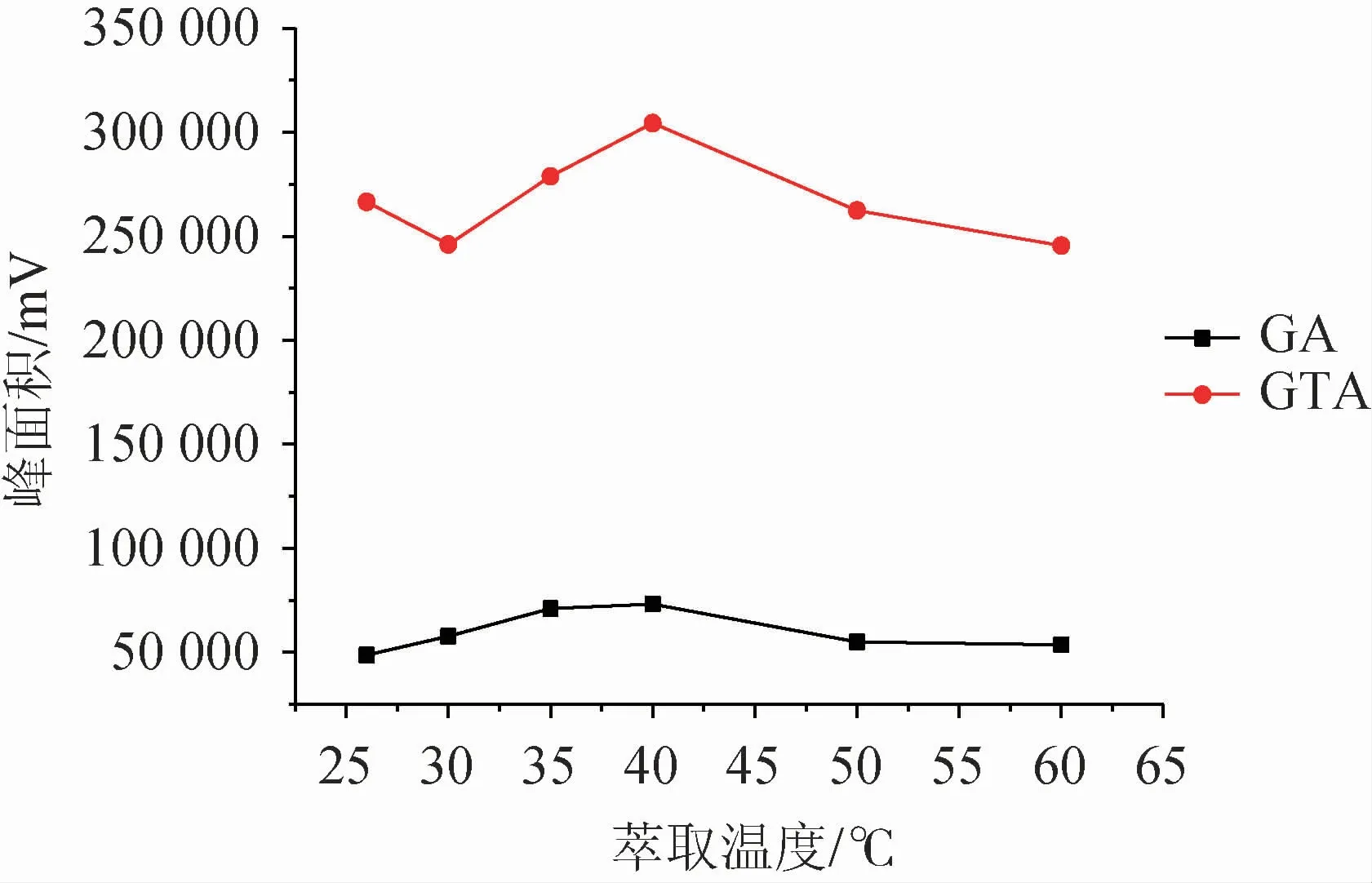
圖5 萃取溫度對萃取效率的影響Fig.5 Effect of extraction temperature on extraction efficiency
2.1.6 萃取時間的選擇
液相微萃取是目標化合物在供相和萃取相之間達到分配平衡的過程,因此對于萃取時間的控制尤為重要。時間過短,萃取未到達平衡,導致萃取不佳;時間過長,同樣也會引起萃取劑的損失。實驗考察了萃取時間5~30 min內對萃取效果的影響,結果見圖6。由圖6可知,隨著萃取時間的增加,萃取效率先增大后降低。當萃取時間為15 min時,甘草酸達到最佳萃取效果;甘草次酸萃取效果在20 min時達到最佳,但在15 min時也達到了較高水平,考慮到整體實驗效率,盡可能在較短的時間完成萃取,最終確定萃取時間為15 min。
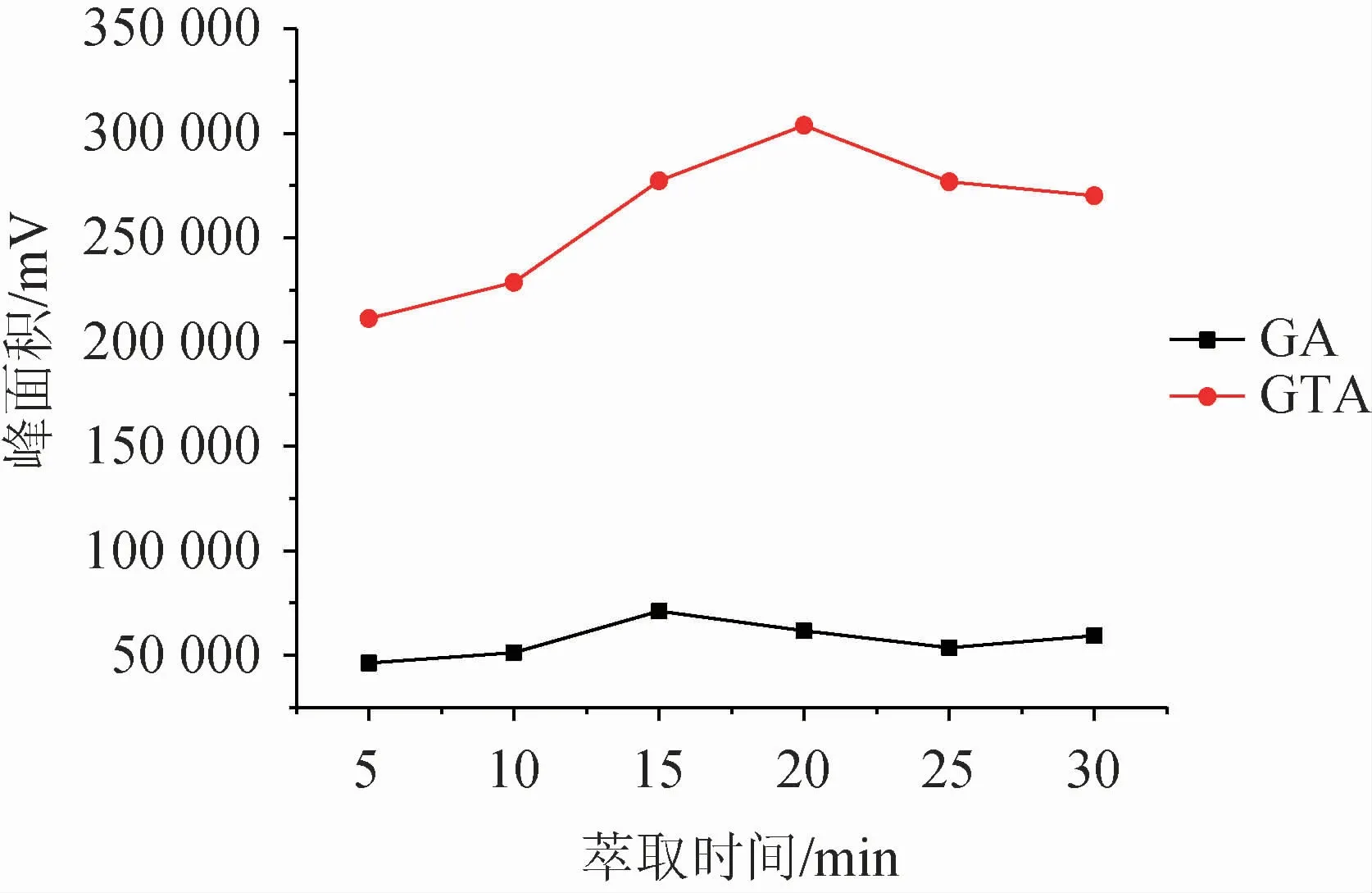
圖6 萃取時間對萃取效果的影響Fig.6 Effect of extraction time on extraction efficiency
2.1.7 驗證試驗
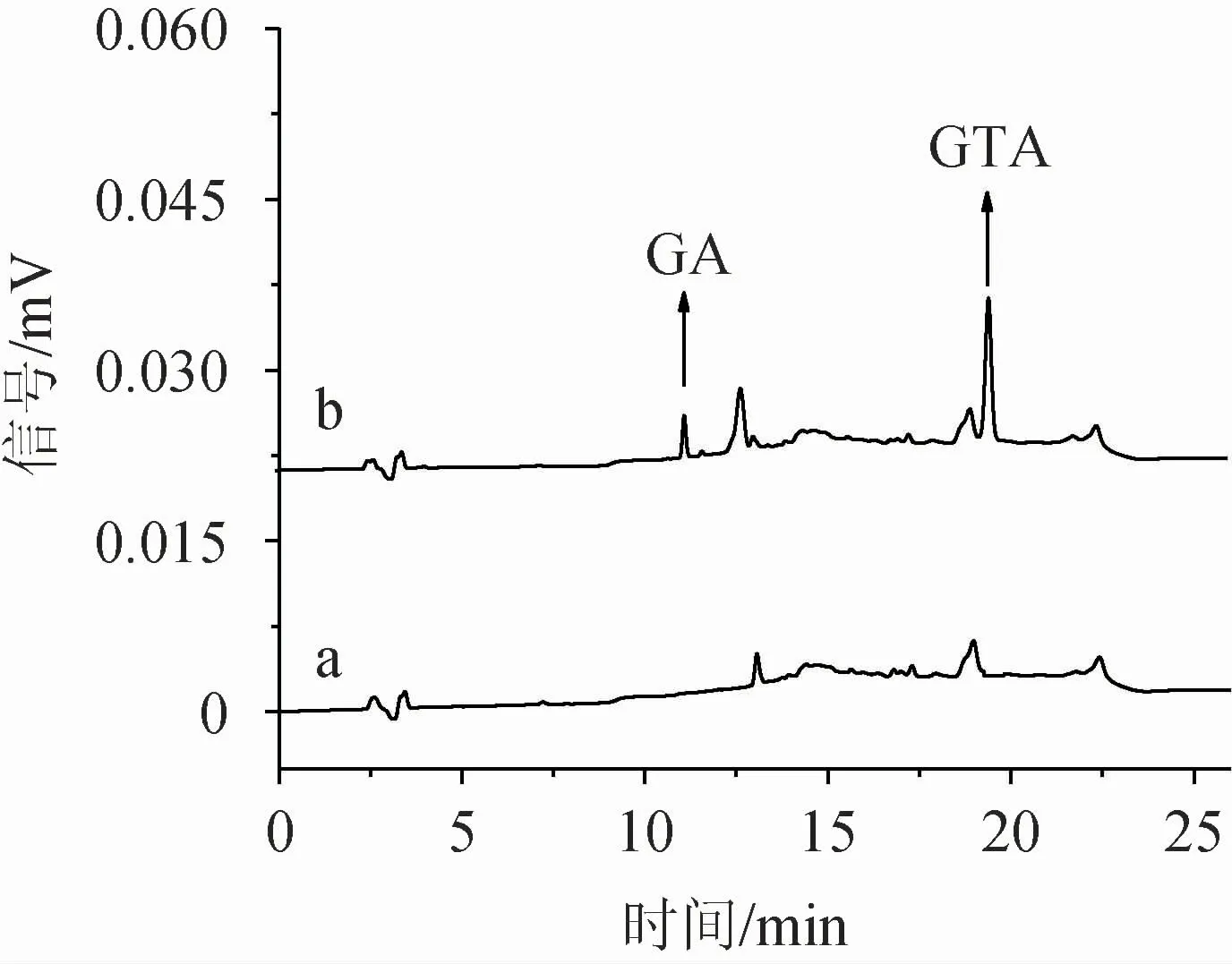
圖7 標準溶液及樣品中甘草酸及甘草次酸色譜圖Fig.7 Chromatograms of glycyrrhizic acid and glycyrrhetinic acid of standard solution and sample
2.2 方法學考察
2.2.1 標準曲線、檢出限及定量限
分別取適量甘草酸、甘草次酸標準儲備液,用水配制成質量濃度為2.50 ng/mL、5.00 ng/mL、10.0 ng/mL、25.0 ng/mL、50.0 ng/mL的標準工作液,而后使用最佳的SFOD-LPME條件進行萃取,并注入色譜系統進行分析。使用甘草酸、甘草次酸工作液萃取后峰面積(y)與質量濃度(x)繪制標準曲線,結果見表2。
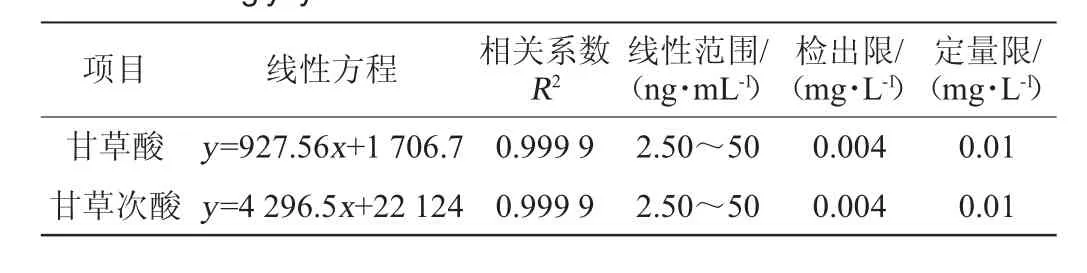
表2 甘草酸和甘草次酸的回歸方程、相關系數、線性范圍、檢測限及定量限Table 2 Regression equation, correlation coefficients, linear ranges,detection limits and quantitation limits of glycyrrhizic acid and glycyrrhetinic acid
由表2可知,在2.50~50.0 ng/mL范圍內,甘草酸、甘草次酸顯示良好的線性關系,相關系數R2均為0.999 9。注入樣品萃取空白,計算信噪比,分別以3倍信噪比及10倍信噪比計算得該方法的檢出限及定量限,得甘草酸、甘草次酸檢出限為0.004 mg/L,定量限為0.01 mg/L,滿足檢測要求。
2.2.2 方法精密度及回收率考察
方法選取清香型、濃香型、醬香型、米香型四種風味的白酒基質進行加標回收試驗,加標量分別為0.01 mg/L、0.05 mg/L、0.40 mg/L的3水平3平行試驗,按照本方法進行萃取分析,加標回收率試驗結果見表3。
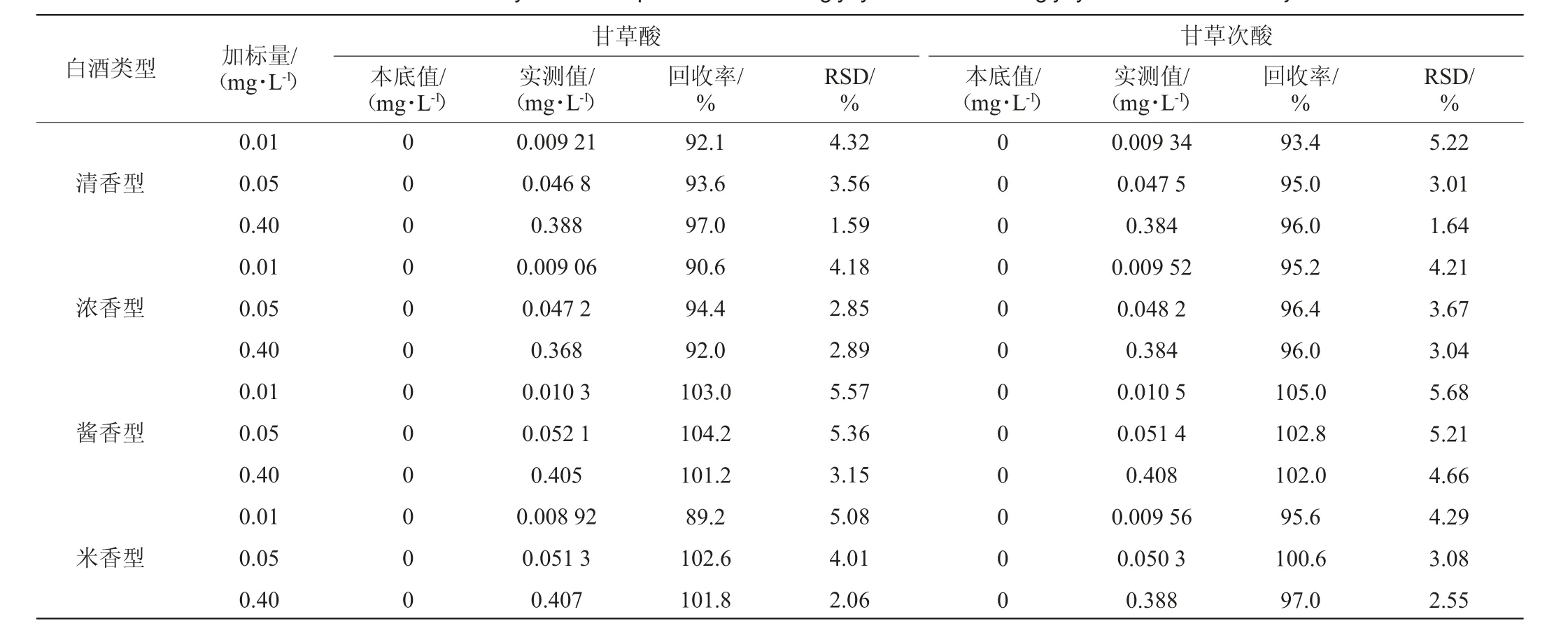
表3 白酒中甘草酸、甘草次酸的加標回收率及精密度試驗結果Table 3 Standard recovery rates and precision tests of glycyrrhizic acid and glycyrrhetinic acid in Baijiu
由表3可知,清香型白酒回收率在92.1%~97.0%之間,精密度試驗結果RSD在1.59%~5.22%之間;濃香型白酒回收率在90.6%~96.4%之間,精密度試驗結果RSD在2.85%~4.21%之間;醬香型白酒回收率在101.2%~105.0%之間,精密度試驗結果RSD在3.15%~5.68%之間;米香型白酒回收率在89.2%~102.6%之間,精密度試驗結果RSD在2.06%~5.08%之間,四種香型白酒基質間加標回收結果均良好,且無明顯差別,說明該方法整體適應性較好,受基質影響小。總體而言,甘草酸加標回收率在89.2%~104.2%之間,甘草次酸加標回收率在93.4%~105.0%之間,精密度試驗結果RSD分別在1.59%~5.57%及1.64%~5.68%之間,精密度和回收率均符合檢測要求。
2.3 實際樣品的測定
對隨機采購的四種香型共50份白酒樣品進行檢測,每份樣品平行測定3次,檢測結果顯示,樣品中均無甘草酸、甘草次酸檢出。
3 結論
隨著市場對天然甜味劑的認可,其替代傳統甜味劑必然是大勢所趨,雖然甘草酸、甘草次酸作為甜味劑添加在食品中不僅能夠改善口感,還具有營養保健的作用,但是對于白酒行業來說,這是對傳統工藝的傷害,更是對消費者的欺瞞,甚至會引發企業間不正當競爭。因此,對白酒中天然甜味劑含量展開監測仍然十分必要。高效天然甜味劑的推廣和使用,將對檢測方法的靈敏度提出更高的要求,同時也是對白酒中甜味劑違法添加的監管提出了一個新的挑戰。本研究建立SFOD-LPME-HPLC法測定白酒中天然甜味劑甘草酸、甘草次酸,方法靈敏度高,操作簡單,富集效果顯著,適用于白酒中微量甘草酸、甘草次酸的分析檢測,可為白酒中添加劑的檢測提供方法參考,有助于建立相關標準及預警機制,對市場形成更有效的監管。
