新生兒多臟器型假性醛固酮減少癥Ⅰ型
2023-07-17 07:17:40曹芯誠陳園園張可張迅捷楊琳李志華
中國當代兒科雜志 2023年7期
曹芯誠 陳園園 張可 張迅捷 楊琳 李志華
(國家兒童醫學中心/復旦大學附屬兒科醫院 1.新生兒科;2.臨床藥學部;3.兒科研究所,上海 201102)
1 前言
假性醛固酮減少癥(pseudohypoaldosteronism,PHA)又稱醛固酮不敏感綜合征,醛固酮水平增高,但因醛固酮抵抗而導致排鉀和排氫減少,臨床以高鉀血癥、代謝性酸中毒為特征,多伴有低鈉血癥。PHA 根據臨床表現和遺傳學特征可分為PHA Ⅰ型、PHA Ⅱ型和PHA Ⅲ型[1]。PHA Ⅱ型又稱戈登綜合征,為常染色體顯性遺傳,以高血鉀、高血氯及代謝性酸中毒和低腎素性高血壓為臨床特征。PHA Ⅲ型又稱繼發性PHA,常繼發于腎臟疾病,往往有腎功能不全。PHA Ⅰ型根據受累器官及基因變異分為腎型PHA Ⅰ和多臟器型PHA Ⅰ(systemic PHA type Ⅰ,sPHA Ⅰ)。腎型PHA Ⅰ為常染色體顯性遺傳,是由腎臟醛固酮受體基因NR3C2突變導致腎小管對醛固酮不敏感,丟失鈉鹽,病變局限于腎臟,表現為低鈉、高鉀、脫水,但病情較輕,隨年齡增長可自行緩解[2]。sPHA Ⅰ是由醛固酮發揮效應的上皮鈉通道(epithelial sodium channel,ENaC)基因突變所致,為常染色體隱性遺傳,發病率僅為1∶166 000[3]。由于ENaC 分布于多個臟器,因此多在新生兒期起病,表現為嚴重電解質紊亂(高鉀血癥、低鈉血癥)和代謝性酸中毒,還可能合并呼吸系統(呼吸窘迫等)、消化系統(嘔吐、拒奶、腹瀉等)和循環系統(心律失常、休克等)等多系統受累[4],治療困難,隨時可因電解質紊亂而危及生命,需終身服藥。本文報道1例以嬰兒失鹽綜合征起病的新生兒sPHA Ⅰ型的多學科診治,為早期診斷、個體化治療及改善這類患兒的長期預后提供經驗。
2 病例介紹
現病史:患兒男,生后18 d,因發現反復血鉀升高10余天轉入我院。患兒系第1胎第1產,剖宮產出生,胎齡38+6周,出生體重3 400 g。出生時羊水清,臍帶、胎盤未見異常,生后Apgar 評分1 min 10分、5 min 10分。患兒生后5 d出現吃奶欠佳,后逐漸加重,出現拒奶、嗜睡、呻吟,生后8 d 至當地醫院就診,考慮“休克、重癥肺炎、呼吸衰竭”收住入院。入院后禁食,并予氣管插管機械通氣、頭孢哌酮舒巴坦鈉抗感染等治療,查血K+11.4 mmol/L(參考值:3.5~5.5 mmol/L),血Na+117 mmol/L(參考值:135~155 mmol/L),pH 7.145(參考值:7.35~7.45),堿剩余-10.1 mmol/L(參考值:-3~3 mmol/L),予補鈉、糾正酸中毒和血液凈化降鉀治療;血清皮質醇45.27 μg/dL(參考值:上午4.26~24.85 μg/dL,下午2.9~17.3 μg/dL),予補充氫化可的松(生后8 d 至轉我院前,每12 h靜脈注射20 mg)和9α-氟氫可的松(生后8 d至轉我院前,每12 h口服0.1 mg)治療。經治療后患兒呼吸平穩、血K+正常(最低3.8 mmol/L),生后11 d 予配方奶喂養,開奶5 d 達全腸內營養后血K+再次升高(最高7.5 mmol/L),再次禁食。為求進一步診治,在鼻導管吸氧下轉入我科。患兒父母均健康,非近親婚配,否認家族遺傳性疾病史。
入院體格檢查:鼻導管吸氧下,體溫36.5°C,心率122次/min,呼吸45次/min,血壓70/40 mmHg(平均壓50 mmHg),體重3 540 g。神志清楚,反應可,全身皮膚紅潤,雙側瞳孔對光反射正常。心律齊,有力,無雜音。雙肺呼吸音對稱。腹部軟,腸鳴音弱。四肢肌張力低,原始反射均未引出,正常男性外生殖器,無陰莖肥大,無色素沉著。
輔助檢查:血氣分析示K+7.0 mmol/L,Na+131 mmol/L, Cl-98 mmol/L ( 參考值: 96~106 mmol/L),pH 7.286,堿剩余-7.1 mmol/L。血肌酐18 μmol/L(參考值:13~33 μmol/L)。血串聯質譜和尿串聯質譜檢測陰性。皮質醇、促腎上腺皮質激素、17α-羥孕酮、游離甲狀腺素和促甲狀腺激素在正常范圍內,醛固酮(>1 000 pg/mL)升高、血管緊張素Ⅱ(278 pg/mL)升高、腎素(>500 pg/mL)升高。B超提示頭顱、雙腎、雙腎上腺、輸尿管和膀胱無明顯異常,腎上腺增強CT 未見明顯異常,心電圖未見明顯異常。入院10 d 后外院家系全外顯子組測序(whole exome sequencing,WES)結果回報檢測到SCNN1A基因復合雜合變異c.723C>G(p.Tyr241Ter, 402)和c.604C>T(p.Arg202Ter, 468)(NM_001038)。
3 多學科診療
3.1 新生兒重癥監護病房初診
該患兒因反復高鉀血癥、低鈉血癥和代謝性酸中毒入院,考慮嬰兒失鹽綜合征。嬰兒失鹽綜合征與鹽皮質激素即醛固酮的分泌及效應有關。醛固酮由腎上腺皮質球狀帶分泌,各種腎上腺皮質功能不全(如先天性腎上腺皮質增生癥、腎上腺發育不良及各種因素導致的腎上腺功能減退)可導致醛固酮分泌不足,不能保鈉排鉀,發生電解質紊亂。而當腎上腺皮質功能正常,但終末器官對醛固酮不應答也會引起低鈉高鉀,如PHA。此患兒外院首先考慮先天性腎上腺皮質增生癥導致醛固酮合成不足,但外院查皮質醇水平并不低,我院進一步送檢激素檢查示醛固酮、血管緊張素Ⅱ、腎素明顯升高,皮質醇、促腎上腺皮質激素在正常范圍內,且腎上腺影像學檢查未見異常增厚或發育不良,給予足劑量足療程的糖皮質激素及鹽皮質激素替代治療后效果不佳,故除外真性醛固酮減少,應考慮醛固酮發揮效應異常,其中又包括腎性及腎外因素導致的失鹽。腎性因素是由于各種腎前性、腎性及腎后性因素導致急性或慢性腎功能不全,影響腎臟排泄鈉鉀功能,該患兒無腎臟基礎病史,雖病程中合并泌尿系感染,但多次監測腎功能及泌尿系超聲結果正常,不支持該原因。腎外因素即PHA,考慮為以終末器官不應答(醛固酮抵抗)為表型的遺傳代謝性疾病,具體分型需待基因結果進一步明確。
3.2 內分泌科會診
該患兒表現為激素替代無效的頑固性電解質紊亂和代謝性酸中毒,醛固酮顯著升高,考慮終末器官對醛固酮無反應,PHA 可能性大。患兒血鈉持續偏低、血壓正常,不支持PHA Ⅱ型。患兒腎功能及腎臟結構正常,雖有泌尿系感染但不足以引起嚴重的電解質紊亂,不支持PHA Ⅲ型,故首先考慮PHA Ⅰ型。該患兒生后起病早,臨床有反復高鉀血癥、失鹽和代謝性酸中毒,還合并脫水、呼吸困難等癥狀,sPHA Ⅰ型應重點考慮,但需待基因報告明確分型。PHA 治療包括降鉀治療(減少鉀攝入和促進鉀排出)、補充鈉鹽和糾酸等對癥支持治療。
3.3 遺傳代謝分子診斷中心會診
對于可治性遺傳病,WES 技術可以幫助制定精準治療方案,以降低病死率、改善預后。該患兒WES結果顯示SCNN1A基因存在復合雜合變異,c.604C>T(p.Arg202Ter, 468)來自父親,c.723C>G(p.Tyr241Ter, 402)來自母親,均未被既往文獻報道,結合該患兒的臨床表型考慮為致病基因變異[5]。患兒明確診斷為sPHA Ⅰ型,為常染色體隱性遺傳,父母雙方均為攜帶者。sPHA Ⅰ型是由于分布于腎小管、呼吸道、皮膚等ENaC亞基基因突變所致[6]。ENaC 是由α、β、γ 亞基形成的多聚體,分別 由SCNN1A(12p13.31)、SCNN1B(16p12.1) 和SCNN1G(16p12.1)編碼[7]。sPHA Ⅰ型常表現為高鉀血癥、低鈉血癥、脫水和代謝性酸中毒,合并呼吸道感染、皮疹和膽汁淤積癥等[4]。
3.4 營養科會診
減少患兒鉀的攝入有利于維持血鉀水平穩定,可考慮以低鉀配方奶粉喂養。常見的低鉀配方奶粉為慢性腎臟病患兒服用的低鉀低蛋白配方奶粉(即腎功能衰竭奶粉),每100 mL 奶液含鉀約22 mg,但同時其他營養成分主要適用于腎功能衰竭嬰兒,不利于該患兒長期的生長發育。母乳含鉀偏低,如果沒有母乳,可適當選擇含鉀偏低的普通一階段配方奶。
3.5 藥劑科會診
考慮患兒需要終身降鉀治療,利用聚磺苯乙烯鈉散(降鉀樹脂)降鉀治療是一種安全、有效、可行的方法。小兒降鉀樹脂的用法包括加入奶中口服和保留灌腸。然而,降鉀樹脂尚無新生兒推薦劑量,用藥期間需長期監測電解質水平,當血K+降到4~5 mmol/L時應考慮停藥。
3.6 新生兒重癥監護病房診斷思路總結
該患兒以嬰兒失鹽綜合征起病,血氣分析提示高鉀血癥、低鈉血癥和代謝性酸中毒,醛固酮水平增高,下丘腦-垂體-腎上腺軸激素水平基本正常,不支持真性醛固酮減少。患兒腎臟結構及功能正常,除外腎性因素導致醛固酮作用受限,考慮醛固酮受體或ENaC基因異常導致的假性醛固酮減少癥,WES 結果也證實該診斷,且明確分型為sPHA Ⅰ型。予降鉀樹脂降鉀、補充鈉鹽治療,并糾正酸中毒,維持內環境穩定。嬰兒失鹽綜合征的鑒別診斷思路見圖1。患兒最終診斷為:新生兒sPHA Ⅰ型、電解質紊亂(高鉀血癥、低鈉血癥)、代謝性酸中毒。
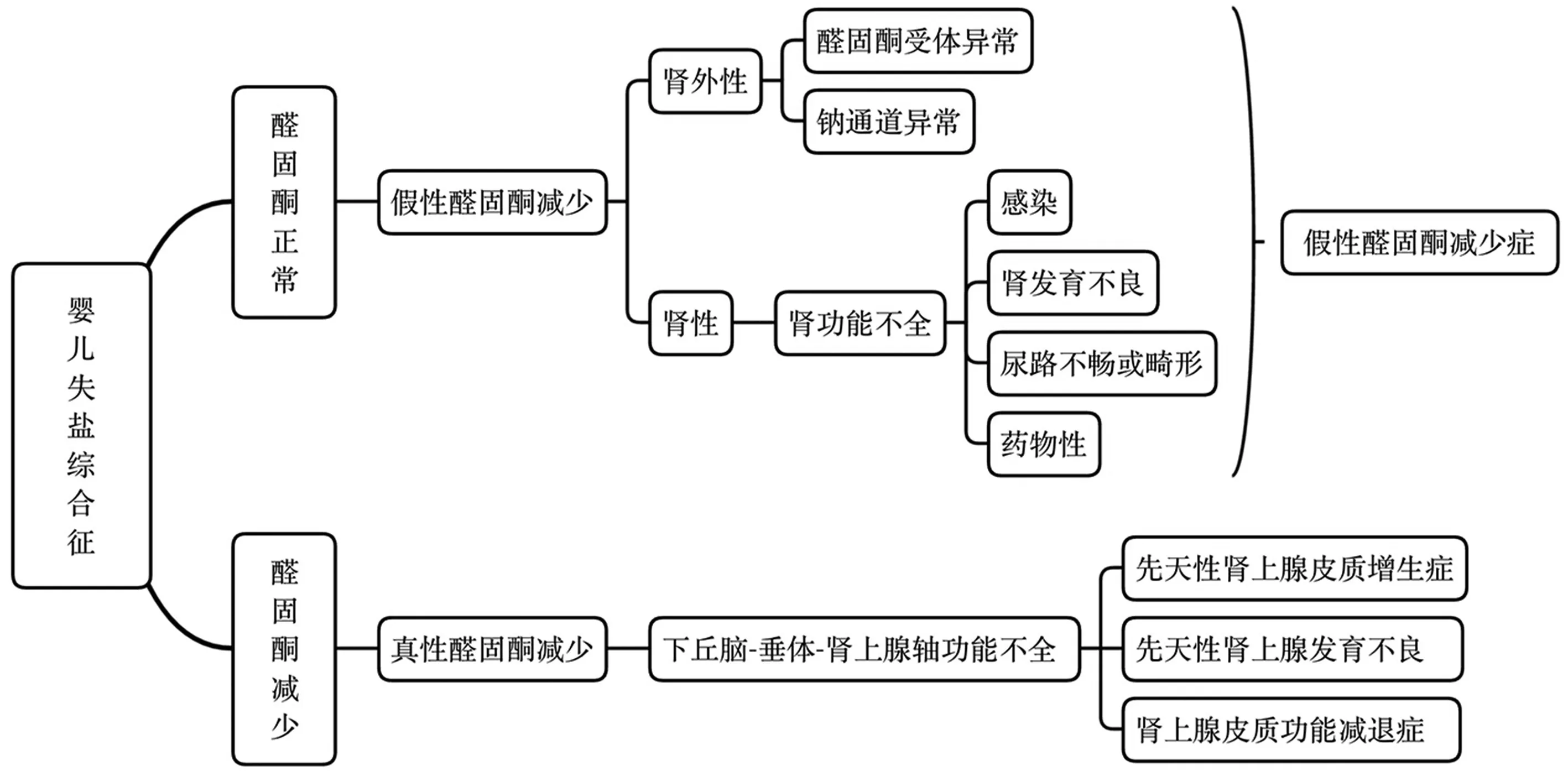
圖1 嬰兒失鹽綜合征鑒別診斷圖
4 住院經過及轉歸
患兒入院時存在頑固性電解質紊亂,予禁食、輸注無鉀和含高鈉的靜脈營養,并予以糾正酸中毒、呋塞米利尿和靜脈滴注胰島素降血鉀。經過內分泌科和遺傳代謝分子診斷中心協診,結合WES結果,明確診斷患兒為sPHA Ⅰ型,逐漸減停糖皮質激素及鹽皮質激素,予對癥補鈉和降鉀治療。降鉀治療包括減少鉀攝入和促進鉀排出,然而,使用常規的促排鉀藥物如胰島素、呋塞米降鉀時血鉀水平波動大;如果長期依賴無鉀靜脈營養患兒也無法出院回家,但含鉀較低的腎功能衰竭奶粉不能滿足患兒正常生長發育需求,因此經過和藥劑科、內分泌科及營養科討論后予降鉀樹脂降血鉀治療。最初考慮到降鉀樹脂分子量大,不易溶解吸收,曾嘗試保留灌腸,但需每次用20 mL注射用水配置降鉀樹脂后經直腸灌入并保留20 min,患兒耐受欠佳且降鉀樹脂易隨大便排出,具體藥量和藥效較難估計,故改為降鉀樹脂每日1 g/kg 分次隨奶液口服,每次先將降鉀樹脂溶于20 mL奶液中形成混懸液喂服保證藥物攝入,再繼續喂奶。同時給予患兒10% NaCl每日15~18 mmol/kg加入奶中口服,基本糾正低鈉血癥,但血鉀仍時有升高。由于含10% NaCl 的奶口味偏咸,患兒常出現拒奶情況,因此嘗試用枸櫞酸鈉來替代10%NaCl口服。使用枸櫞酸鈉治療后患兒血鉀也降至正常水平,降鉀樹脂的用量減少,考慮可能枸櫞酸鈉偏堿,而堿性環境也利于降鉀。最終患兒體重4 410 g,使用一階段配方奶880 mL/d,補鈉量為枸櫞酸鈉6.48 mmol/(kg·d)和10% NaCl 6.17 mmol/(kg·d),降鉀樹脂2 g/d,電解質水平保持穩定,好轉出院。
其他方面治療如下。(1)呼吸:患兒入院時有呼吸急促,予低流量鼻導管吸氧3 d;(2)感染:患兒住院期間1月齡時發生肺炎克雷伯桿菌尿路感染,根據藥敏試驗結果予頭孢吡肟抗感染后很快治愈。為了患兒出院后的長期管理,出院前我們教會患兒父母計算每日口服的奶液及各種藥物中K+和Na+的含量并做好記錄,指導家長帶患兒定期前往當地婦幼保健院監測血氣及電解質,并根據隨訪結果調整藥物劑量。患兒出院返家后曾有1次呼吸道感染,隨訪至2023年5月時患兒4月齡,發育正常,抬頭穩,咿呀學語,愛笑,體重達7.1 kg(P50~P75),每日奶量達1 050 mL,每日口服10%NaCl 21 mL、枸櫞酸鈉77 mL,奶及藥物中含鈉量合計17.6 mmol/(kg·d);每日口服降鉀樹脂2.5 g,每周監測1 次血氣電解質,確保電解質水平保持穩定。
5 小結
本文報道了1例新生兒期以嬰兒失鹽綜合征起病的sPHA Ⅰ型,基于新生兒科初診的鑒別診斷,與內分泌科、遺傳代謝分子診斷中心、營養科和藥劑科開展多學科診療合作進行診斷和救治。新生兒期起病的嬰兒失鹽綜合征臨床表現常缺乏特異性,需排查下丘腦-垂體-腎上腺軸功能(醛固酮、皮質醇、促腎上腺皮質激素、腎素、血管緊張素等)明確有無醛固酮產生不足,進而排查腎臟疾病除外腎性因素導致醛固酮作用障礙,之后考慮醛固酮受體或ENaC異常導致醛固酮不敏感的原發性PHA,并借助基因檢測技術明確診斷及分型[8]。
sPHA Ⅰ型患兒易發生嚴重電解質紊亂,治療上需口服鈉鹽促進鈉鉀代謝,并輔以降鉀樹脂維持鉀離子在正常范圍內,且需終身治療并定期監測電解質水平,及時調整口服藥物劑量,充分告知家長疾病的風險及長期監測的必要性,教會家長學會自我管理及隨訪就醫指征。研究報道,近50%的sPHA Ⅰ型患兒在長期補鈉及口服降鉀樹脂下病情保持平穩,約10%患兒死于因高鉀血癥出現的心律失常[9]。該患兒確診后,給予小劑量降鉀樹脂及口服枸櫞酸鈉和10% NaCl 治療,監測血鉀和血鈉水平保持穩定,達臨床痊愈出院,在門診長期隨訪。sPHA Ⅰ型作為較少見的引起新生兒期嚴重電解質紊亂的疾病,需在早期識別與診斷后盡早開始降鉀補鈉及對癥治療,對于減輕多臟器損傷至關重要。
利益沖突聲明:所有作者聲明無利益沖突。
