南充地區脊髓性肌萎縮癥攜帶者篩查及高風險胎兒產前診斷分析
2023-06-27 08:43:02陳麗平何勇均蔡燕石琪
川北醫學院學報 2023年6期
陳麗平,何勇均,蔡燕,石琪
(川北醫學院附屬醫院,1.遺傳與產前診斷中心;2.婦產科,四川 南充 637000)
脊髓性肌萎縮癥(spinal muscular atrophy,SMA)是一種嚴重的常染色體隱性遺傳性神經肌肉疾病,主要是位于5q13.2區域的運動神經元存活基因1(survival motor neuron,SMN1)致病性變異所致,當SMN1基因第7或第7、8外顯子發生純合缺失或點突變時引起SMN蛋白表達缺失,造成脊髓前角α運動神經元變性,導致近端肢體和軀干進行性、對稱性肌無力和肌萎縮[1]。該病在新生兒中的發病率約為1/10 000[2],攜帶者頻率為1/50~1/40。兒童型SMA(1~3型)的患病率為2.78~6.56人/10萬人[3],其中4型SMA患病率約為0.32人/10萬人[4]。SMA居<2歲兒童致死性遺傳病首位,其中累及呼吸系統導致的呼吸衰竭是最常見的致死原因[5]。中國人群中的攜帶者頻率約為1/42[6],但是各地區尚無發病率的確切數據。
SMA臨床表現嚴重,人群中的攜帶率較高,且致病基因明確,2008年美國醫學遺傳學與基因組學學會(ACMG)[7]建議無論地域種族,應該對所有育齡人群進行SMA攜帶者篩查。目前已報道的SMA攜帶者篩查技術眾多,根據SMA致病基因的特點,對SMN1基因外顯子7、8缺失進行檢測,已經成為SMA攜帶者篩查的首選方法[8]。目前尚無關于南充地區人群的SMA攜帶率的相關研究,本研究從此角度出發,采用多重連接依賴性探針擴增(MLPA)技術和熒光定量聚合酶鏈式反應(qPCR)技術對南充地區4 325例因不良妊娠史就醫的育齡期女性及183名配偶的SMN1基因進行檢測,調查SMA攜帶率,為本地區的SMA的篩查及孕婦的產前診斷提供一定的指導。
1 資料和方法
1.1 一般資料
選取2019年12月至2022年10月川北醫學院附屬醫院收治的4 325例定期產檢且因不良妊娠史就醫的育齡期女性及183名配偶為研究對象,其中孕婦年齡(29.35±2.17)歲;孕周9~20周。本研究獲得醫院倫理委員會的批準,所有納入的研究對象均進行檢測前溝通,取得研究對象的知情同意并簽署相關知情同意書。
1.2 方法
1.2.1 樣本采集 所有受檢者均進行外周靜脈血采集2 mL,采集后的血液進行EDTA抗凝,于4 ℃的環境中保存,對采集的血液樣本均在1周內完成基因檢測。
1.2.2 DNA提取 采用達安基因NP986-C系統進行外周血核酸的提取,采用NanoDropOne超微量生物檢測儀進行DNA濃度檢測。
1.2.3 基因檢測 采用熒光定量PCR法對SMN1基因進行檢測,檢測試劑盒購自上海五色石公司,操作方案嚴格按照試劑盒說明書進行。PCR擴增條件為:95 ℃預變性(10 min)→95 ℃變性(15 s)→58 ℃退火延伸(60 s),共40個循環。采用FAM及VIC通道進行信號采集。若檢測孕婦為SMA攜帶者,則對其配偶進行樣本采集及檢測,配偶檢測方法同上。若配偶檢出為SMA攜帶者,則進行胎兒的基因檢測,經夫婦雙方知情同意后,于妊娠的16~22周進行經腹壁羊膜腔穿刺術(B超引導下),用一次性無菌注射器緩慢抽取孕婦羊水10 mL,利用購自荷蘭MRC-Holland公司的MLPA P060試劑盒(SALSA MLPA Kit P060-B2)經變性、雜交、連接以及擴增后,產物通過ABI3500基因分析儀(ThermFisher,美國)檢測,驗證SMN1基因拷貝數變異,數據使用Coffalyser V8.0軟件分析。
2 結果
本研究納入4 508例研究對象(育齡女性4 325例及部分女性的配偶183例),共檢測到SMA攜帶者70例,攜帶率為1/64.4,其中E7和E8雙位點雜合缺少的64例(1/70.4),E7位點雜合缺失6例(1/751.3)。對夫妻雙方均為雜合性缺失胎兒的基因檢測發現其為SMN1基因E7位點純合缺失。基因拷貝數變異檢測發現,胎兒SMN1基因為0個拷貝數,SMN2基因為3個拷貝數。將胎兒預后詳細告知胎兒父母后,胎兒父母決定終止妊娠。見圖1。
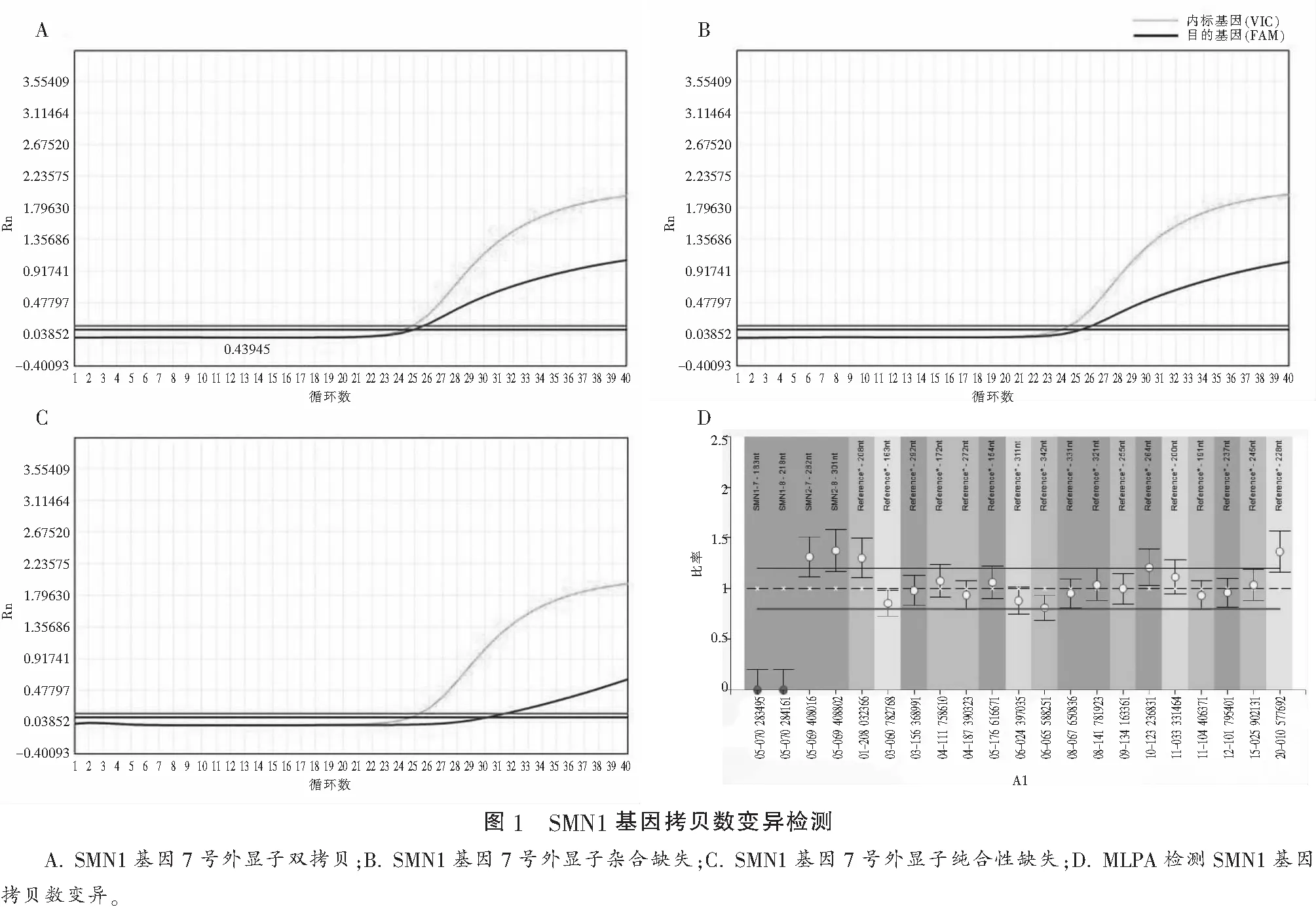
3 討論
SMA是遺傳性神經肌肉疾病的一種,主要因為脊髓前角細胞和低位腦干運動神經核團變性,導致不同程度的肌張力降低、肌萎縮,嚴重時可影響呼吸肌危及生命。該病常呈家族性,從胎兒期到成人不同年齡均可發病,且治療方法有限,預后通常不佳。SMA治療的研究領域近年來發展迅速,部分相關治療藥物已經成功獲批上市,如索伐瑞韋、利司撲蘭、諾西那生等[9],可從不同方面提高全長SMN蛋白水平,其他治療方式(如神經保護治療、SMN修飾基因治療、肌肉激活治療、干細胞治療等)也相繼取得了進展。上述治療給SMA患兒帶來了希望,但早診斷、早治療極其重要[10]。目前藥物必須依靠進口,售價高昂,普通家庭難以承擔,因此,利用先進的技術對孕婦SMA攜帶率的篩查,預防SMA患兒的出生,是一種經濟且有效的SMA防治方法,對減輕家庭的經濟負擔和社會的負擔均有較好的價值。
對育齡期女性SMA攜帶情況進行篩查屬于一級預防,不僅能避免SMA患兒的出生,而且還能夠提高我國新生兒的人口質量。臺灣地區在2005至2009年共計檢測了10萬余例孕婦,其中共檢測出SMA攜帶者2 262例,攜帶率約為1∶48[11];2013年我國上海地區同樣對4 719例孕婦進行了篩查,SMA攜帶率約為1∶52[12];隨后相繼有廣西、云南及陜西地區報道了關于本地區的SMA篩查結果,其中以廣西地區的攜帶率較低,約為1∶81[13]。目前尚沒有南充市及周邊地區關于SMA攜帶率的研究,本研究納入4 508例研究對象(孕婦4 325例及部分配偶183例),共檢測到SMA攜帶者70例,攜帶率為1/64.4,其中E7和E8雙位點雜合缺少的64例(1/70.4),E7位點雜合缺失6例(1/751.3),基本符合當前我國的SMA攜帶者的流行病學調查結果。同時對夫婦均為SMA攜帶者的胎兒基因檢測發現其為SMN1基因E7位點純合缺失。MLPA技術檢測發現,胎兒SMN1基因為0拷貝,SMN2基因為3個拷貝數,最終通過遺傳咨詢預防SMA患兒的出生。
SMA患兒的父母多為SMN1單拷貝攜帶者,但也約有4%的SMA攜帶者為兩個SMN1拷貝位于同一染色體上,這種“2+0”型攜帶者目前缺少準確的檢測技術,需要聯合SMA患者父母的SMN1基因拷貝數和家族史調查來進行診斷評估。同時,現階段對于家庭中有“2+0”類型攜帶者的夫妻,其再發風險和產前診斷策略缺少準確的檢測技術,對于“2+0”類型攜帶者進行病例篩查與遺傳咨詢尤為關鍵[14]。目前臨床上常用的技術是MLPA和熒光定量PCR,但因各自的缺點限制了其在大規模篩查中的應用。已有大量的研究數據表明,高通量測序(NGS)可以直接計算SMN1拷貝數,但篩查或診斷SMN1微小變異仍存在很大困難,目前在我國尚未成為SMA的常規檢測方法。數字PCR(dPCR)法、高分辨率熔融分析法等新興技術方法在臨床SMA攜帶者篩查中有巨大的潛力。但由于dPCR法不能檢測出SMN1基因內點突變以及沉默的SMA攜帶者(2+0),未來需要研究開發可以檢測單核苷酸變化的dPCR技術,以適用于大規模的篩查[15]。
綜上,本研究初步確定了南充地區SMA的攜帶率,同時對同為SMA攜帶者夫妻的胎兒進行了SMN1基因的檢測,最終確定為SMA患兒并避免了此例患兒的出生,減輕了家庭及社會的負擔。本研究也存在有一定的缺陷,如樣本數目少,樣本中配偶人數偏少,未進行SMN2基因的檢測,因此可能會對本研究的結論造成一定的影響,后續有待研究的進一步完善。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
