結合基因關聯和轉錄組分析鑒定小麥成株期抗條銹病位點YrZ501-2BL的候選基因
2023-06-27 08:08:56張旭韓金妤李晨晨張丹丹吳啟蒙劉勝杰焦韓軒黃碩李春蓮王長發曾慶東康振生韓德俊吳建輝
中國農業科學 2023年8期
張旭,韓金妤,李晨晨,張丹丹,吳啟蒙,劉勝杰,焦韓軒,黃碩,李春蓮,王長發,曾慶東,康振生,韓德俊,吳建輝
結合基因關聯和轉錄組分析鑒定小麥成株期抗條銹病位點的候選基因

1西北農林科技大學農學院/旱區作物逆境生物學國家重點實驗室,陜西楊凌 712100;2西北農林科技大學植物保護學院/旱區作物逆境生物學國家重點實驗室,陜西楊凌 712100
【目的】小麥條銹病是小麥的主要病害之一,每年都會對小麥產量安全造成嚴重危害,挖掘小麥抗條銹病基因,為小麥抗條銹病種質創新和揭示小麥抗條銹病遺傳機制奠定基礎。【方法】利用多組學手段結合全基因關聯分析(GWAS)開展對小麥成株期抗條銹病性狀的解析。首先對411份來自CIMMYT和ICARDA的春小麥進行全基因組關聯分析,在小麥2BL染色體上定位到一個主效的成株期抗條銹病位點,并利用含有該位點的抗病材料Z501及感病親本晉麥79的雙親群體進行連鎖作圖,成功驗證了該位點抗性的穩定性,暫命名為。在此基礎上,通過基因注釋、比較基因組分析、轉錄組分析和候選基因的關聯分析對目標區間篩選候選基因。【結果】綜合GWAS和連鎖作圖結果,將鎖定在小麥2B染色體0.26 Mb(575.706—576.587 Mb)范圍內,根據中國春參考基因組注釋信息分析,該區間含有12個基因,其中,高可信基因6個;利用在線網站,將目標區間所在的中國春參考基因組與其他已公布的不同倍性小麥基因組進行比較,發現該區間的6個高可信小麥基因基本都能在其他小麥材料中找到同源基因,且基因排列順序相同,說明該區間可能不存在較大片段的插入、缺失、倒位等現象,可以利用參考基因組信息進行候選基因預測;結合抗病親本Z501和感病親本晉麥79的成株期接種條銹菌后的轉錄組數據,發現只有、和受誘導表達,且抗感病親本存在表達差異。根據中國春基因組注釋,三者分別編碼GATA轉錄因子、SH3P2蛋白和鋅指蛋白。通過進一步的候選基因關聯分析,發現只有SH3P2蛋白中存在與條銹病表型顯著性差異的SNP位點(G/A),雖然該位點(G1369A)在2個可變剪切的轉錄本中均未引起氨基酸編碼變化(TCG和TCA均編碼絲氨酸),但可能與可變剪切有關,同時該位點(G1369A)的不同單倍型表型之間也存在極顯著差異,進一步分析發現,在G1369A位點下游還有2個引起氨基酸改變的變異位點G1377A和G1431A,分別引起纈氨酸(GTT)到異亮氨酸(ATT)和纈氨酸(GTG)到甲硫氨酸(ATG)的改變,但這兩個位點在455份重測序材料中所占比例只有0.87%,屬于稀有變異,因此,未進行顯著性檢驗。綜上,推測為的重要抗病候選基因;此外,針對候選區間的差異SNP開發了相應的AQP標記,可用于輔助選擇,為下一步小麥抗銹病分子育種應用提供了標記資源。【結論】利用多組學整合關聯分析的方法,成功在小麥2B染色體上挖掘到1個抗條銹病候選基因。
小麥;條銹病抗性;;多組學整合分析;SH3P2蛋白
0 引言
【研究意義】小麥條銹病是世界性流行的活體寄生真菌病害,一直是威脅中國小麥安全生產的重要病害。近年來產生的新毒性小種條中34大面積流行,導致當前中國有效的抗條銹病資源極度缺乏,是抗病育種面臨的重要問題[1-2]。因此,快速高效地挖掘小麥重要廣譜抗性資源,克隆其關鍵抗病基因是突破這一瓶頸的前提條件[3]。【前人研究進展】隨著小麥條銹病致病機理的不斷深入探究,發現成株期抗病性往往具有持久性特點,目前,克隆到的成株期抗病基因(如、和)均具有廣譜、抗多種病害及持久的特點[4]。其中,編碼ABC轉運蛋白(ATP binding cassette transporter),通過調控磷脂代謝影響細胞膜結構以及脫落酸信號通路來影響病原菌的生長,達到抗病的目的[5-6];編碼一個包含激酶和脂綁定結構域的蛋白(START-kinase),在相對高溫(25—35℃)條件下,表現出對多個條銹菌小種的廣譜抗性[7-8];編碼己糖轉運蛋白(hexose transporter),通過位點突變產生顯性負效應作用,引起Yr46蛋白喪失對葡萄糖的轉運活性,導致入侵的病原菌不能獲得足夠的糖源,從而使植物表現為抗病[9]。克隆的這3個小麥成株抗性基因,雖抗病機制不同,但均參與能量代謝或糖轉運,與衰老有關,且相對保守[4],3個基因在中國春基因組中均具有相對完整的基因結構。受此啟發,通過對中國春基因組目標區間內基因進行解析,可能會發現與成株期抗條銹病相關的候選基因[10]。隨著小麥參考基因組精細圖譜組裝的完成以及二代測序、組裝和捕獲測序的快速發展[11],相繼開發出一系列基因克隆的新策略,加快了傳統的基因克隆進程[12]。此外,前人開展了大量的全基因組關聯分析(genome-wide association study,GWAS)模型、方法的探索,如日本學者通過全基因組測序,在水稻中開發了一種高效GWAS分析方法,基于對每個核苷酸多態性的功能重要性進行評估,可以快速鑒定候選基因,而不需要額外的試驗[13]。同樣,在小麥中也開展了類似研究,如利用抗病R基因富集測序結合關聯分析開展的小麥稈銹病R基因快速克隆[14],利用外顯子捕獲測序結合關聯分析快速檢測小麥抗葉銹病基因的功能SNP變異[15],以及利用高密度SNP芯片結合關聯分析開展候選基因的發掘與功能驗證等[16-19],使研究者能夠利用多組學手段結合GWAS開展對小麥復雜性狀的解析。國際玉米小麥改良中心(CIMMYT)和國際干旱農業研究中心(ICARDA)的春小麥材料對小麥條銹病具有良好的抗性,前期通過大規模基因組關聯分析在小麥2BL染色體快速發掘4個抗條銹病QTL位點[10],其中,和均已被成功驗證[10, 20]。【本研究切入點】雖然部分位點已經過驗證,但其他興趣目標區間(如第三個主效位點)遺傳基礎還不清楚,特別是與抗條銹病有關的候選基因分析方面,尚未進行深入解析。【擬解決的關鍵問題】本研究著眼于第三個主效位點,利用連鎖作圖、候選基因關聯分析、轉錄組分析等多種手段對目標區間內的候選基因進行快速篩查,確定與抗條銹病有關的關鍵候選基因,為小麥抗條銹病種質創新和揭示小麥抗條銹病遺傳機制奠定基礎。
1 材料與方法
1.1 試驗材料和遺傳群體創制
全基因組關聯分析材料:實驗室前期收集了411份主要來自國際玉米小麥改良中心(CIMMYT)和國際干旱農業研究中心(ICARDA)的春小麥高代品系材料(電子附表1),其中202份ICARDA材料由西北農林科技大學農學院宋衛寧教授惠贈[21],該套材料已完成了在中國條銹病常發區的多年多點的條銹病表型鑒定[21],并進行小麥660K芯片的基因分型,進一步完成了小麥條銹病的GWAS定位工作,在小麥染色體2BL上定位多個抗病QTL,其中包含[10]。
連鎖分析:通過GWAS分析,上述411份小麥材料中,來自澳大利亞抗病品種Z501(PI 410902,系譜為IRN-62-101/Cheyenne)可能含有目標位點,為驗證該位點的準確性及穩定性,以感病品種晉麥79作為受體母本,Z501為供體父本,雜交構建306個F2和F2:3遺傳分析群體,并構建連鎖圖譜進行基因定位。
候選基因關聯分析:實驗室前期收集了455份已進行基因組重測序的材料(其中絕大部分已公開發表)[22-24],該套材料包含農家種、中國歷年審定品種、國外引進品種,以及高代品系等,部分材料由山西農業大學鄭軍副教授惠贈。關于該套材料的詳細信息請見電子附表1。
感病對照品種為銘賢169(MX169)和小偃22(XY22)。
1.2 全基因組關聯分析
利用GEMMA軟件中的單變量線性混合模型對411份小麥材料的成株期表型(實驗室已有多年多點表型數據)及基因型進行全基因組關聯分析[10]。其中,e表示用GEC(Genetic Type I Error Calculator)軟件[25]計算的有效SNP位點數目。利用GEC軟件計算各染色體建議值的范圍為1.36×10-4—9.9×10-4,所以整體考慮3.40×10-4作為顯著性位點的閾值。選擇R軟件的QQman軟件包進行繪制曼哈頓圖[26],采用GCTA軟件[27]計算位點染色體的遺傳率。
1.3 田間試驗及遺傳分析
2017—2018年和2018—2019年播種季在陜西省楊凌西北農林科技大學試驗田種植親本及306個F2單株和F2:3家系。F2:3家系每個家系行長1 m,行距0.2 m,每隔20個家系種植2行小偃22作為感病對照,周邊種植銘賢169作為誘發行。試驗田采取人工接種手段,使用的小麥條銹菌為CYR32、CYR33、CYR34(1﹕1﹕1)。
當感病對照品種銘賢169和小偃22葉片發病面積>80%時,開始對遺傳群體進行表型鑒定。每隔3 d重復一次鑒定,共調查3次,采用反應型(infection type,IT)[28]為鑒定標準,取最高反應型為最終抗病性結果。對于純合抗病/感病家系,只記錄一個表型數值;對于表型分離家系,記錄2個表型數值,即最小值和最大值。在對該群體進行遺傳分析時,利用Excel軟件進行統計分析,參考IT數據,其中,0—3為高抗反應型,4—6為中抗反應型,7—9為感病反應型[29]。
1.4 混池測序(BSA)與基因分型
根據F2和F2:3家系抗條銹病鑒定結果,挑選20個純合抗病家系(IT=0—3)和20個純合感病家系(IT=9)構建抗感池,將雙親與抗感池送至北京博奧生物技術有限公司進行小麥660K SNP芯片的全基因組掃描,分析在抗感池和雙親之間多態性SNP位點在染色體上的分布。利用Excel軟件IF函數篩選抗病親本、感病親本以及抗感池之間的純合差異SNP位點,并參照整合的遺傳圖譜獲得差異SNP位點的染色體分布和物理位置信息[18]。隨后,使用在線設計平臺網站PolyMarker(http://www.polymarker.info/)對所有純合位點的SNP進行等位基因特異的定量PCR基因分型系統(allele-specific quantitative PCR based genotyping assay,AQP)標記[30]設計,再挑選出染色體特異性的標記,根據SNP的物理參考位置及分布密度選取若干AQP標記送公司合成引物。用親本、抗感池和隨機挑選的小樣本群體驗證標記的特異性,將篩選合格的多態性標記用于作圖群體進行基因分型。
1.5 遺傳連鎖圖譜構建及基因定位
各群體的分離比例通過卡方(χ2)分析確定實際值與期望值的顯著性,利用JOINMAP 4.0軟件[31]進行分子標記與條銹病表型之間的連鎖分析,LOD值設為3.0。采用Kosambi函數[32]計算遺傳距離。采用軟件Mapchart V2.3[33]繪制遺傳圖譜。
1.6 比較基因組分析
根據定位結果確定目標位點兩翼最近的標記在中國春參考基因組(IWGSC RefSeqv 1.1)[34]對應的物理位置,在開放網站http://wheat.cau.edu.cn/TGT/上[35],利用10+Genome等基因組信息,進行比較基因組分析,用以觀察目標區間的共線性、染色體結構是否發生變異等情況。
1.7 候選基因預測
目前,小麥族多組學網站(Wheatomics,http://wheatomics.sdau.edu.cn/)[36]整合了已發表的多個小麥族物種的基因組、轉錄組、變異組等多組學數據。根據GWAS和遺傳定位的整合結果,在該網站中獲取目標區間兩端最近的側翼標記之間的中國春IWGSC RefSeq v1.1參考基因組序列,并根據區間內部指定基因的ID獲取對應的基因序列,結合實驗室前期針對Z501和晉麥79的轉錄組數據,利用基因組瀏覽器(JBrowse)對目標區間所有鑒定到的基因進行以下3個層面的分析:1)表達的時空特異性,比較候選基因在不同發育時期、不同材料之間的表達差異;2)mRNA水平是否存在多態性,主要比較候選基因在感病和抗病材料之間是否存在序列變異,這些變異是否導致無義、錯義、移碼突變或剪切模式的改變;3)表達量的變化,利用RNA-Seq數據確認候選基因在不同材料或組織中表達量的相對差異。
1.8 候選基因的關聯分析
調取候選基因在455份普通小麥上的重測序數據,所得序列需覆蓋基因的外顯子、內含子、5′-和3′-非翻譯區、啟動子區。通過與參考基因組中國春序列進行比對,分析核苷酸多態性,包括SNP和InDels。在此基礎上,結合小麥成株期抗條銹病表型,應用GAPIT軟件[37]進行關聯分析,鑒定與抗病表型顯著關聯的功能性遺傳變異及抗病優異單倍型,并繪制LD-map。
針對候選區域內,基于以下原則對核酸多樣性進行分類[13]:G1:引起氨基酸編碼變化和可變剪切位點的顯著關聯SNP;G2:位于啟動子區(起始密碼子ATG上游2 kb)內顯著關聯SNP;G3:位于編碼區的同義突變,內含子區、3′ UTR區顯著關聯的SNP;G4:編碼區(coding regions)外的顯著關聯SNP;G5:不顯著關聯的SNP。
2 結果
2.1 全基因組關聯分析
在單變量線性混合模型分析的基礎上,對411份材料在多年多個田間環境下的條銹病表型(IT和DS值)和最佳線性無偏預測(BLUP)進行關聯檢驗[10]。為方便表述,本文只列出DS-BLUP的GWAS結果(圖1-a),在2B染色體長臂上發現極為顯著的峰值,進一步分析,在該2BL上共發現4個QTL位點,其中,和均在前期已被本課題組驗證[10, 20]。本研究著眼于第3個主效位點(圖1-b),通過整合多個環境的GWAS結果,發現絕大部分顯著性SNP位點都集中在575.706—576.587 Mb范圍內,因此,推斷該區間為的重要目標區間(圖1-c和圖1-g)。
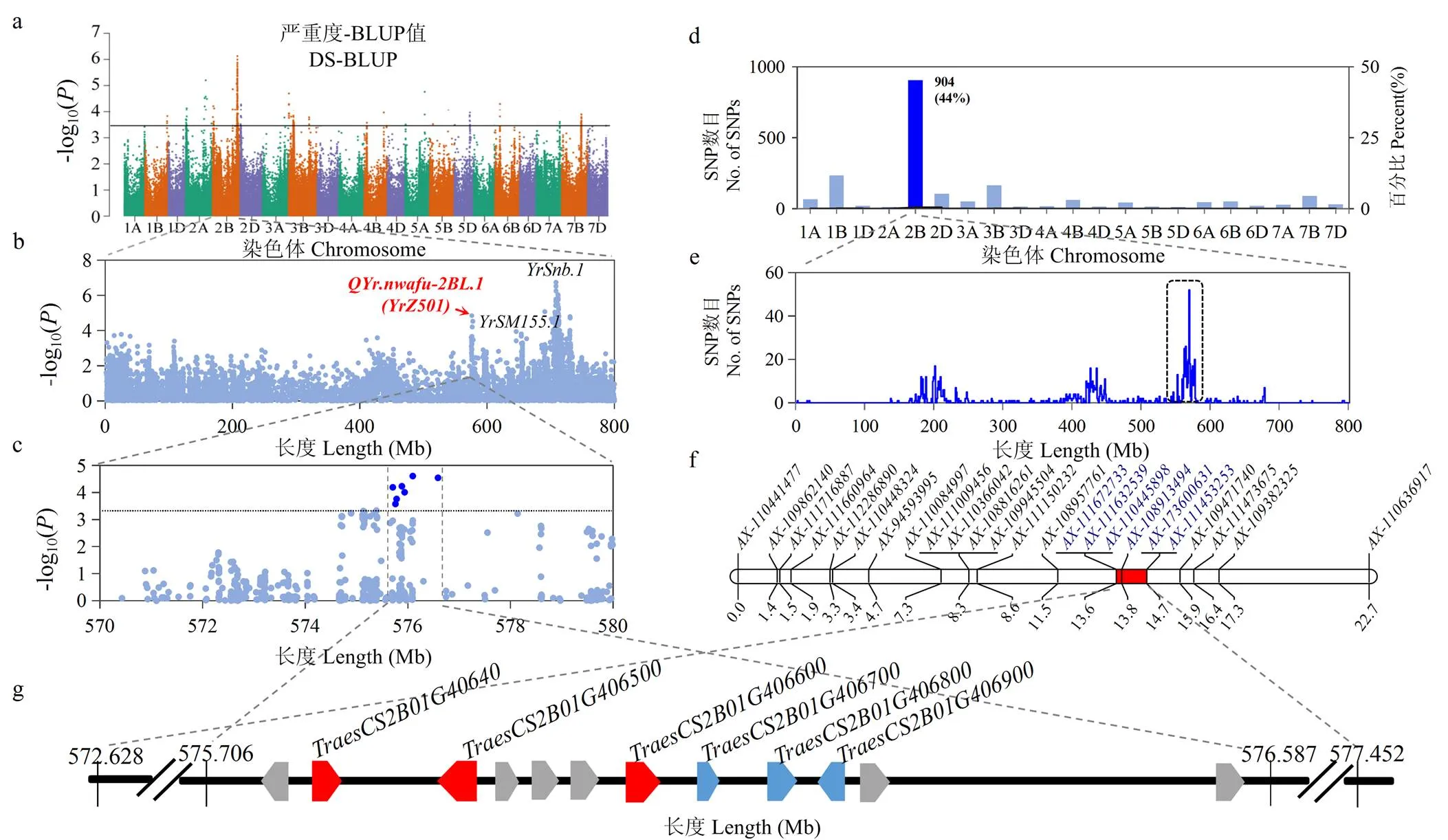
a:利用411份春小麥品系進行條銹病表型GWAS分析,全基因組顯著閾值-log10(P)為3.4(c同理);b、c:2B染色體峰周圍的基于單核苷酸多態性關聯的曼哈頓局部圖,灰色虛線包圍的區域代表潛在的候選區域;d:BSA+660K SNP芯片中差異的多態SNPs在小麥每條染色體上的分布情況;e:多態SNP在2B染色體上的物理位置分布密度;f:基于基因型數據的小麥2B染色體上QYr.nwafu-2BL.1(YrZ501)位點的遺傳連鎖圖譜,紅色條表示QYr.nwafu-2BL.1(YrZ501)位點的候選區間;g:QYr.nwafu-2BL.1(YrZ501)候選區間內的預測基因
2.2 晉麥79×Z501的群體抗病遺傳分析
為進一步確認抗病位點的準確性,根據GWAS結果,找到含有該位點的小麥材料Z501,并對晉麥79×Z501的親本及其后代F2植株和F2:3家系進行表型統計分析。抗病親本Z501表現為高抗(IT=1—3),感病親本晉麥79表現為高感(IT=9)(圖2)。根據F2單株的反應型看,抗病單株67個,感病單株239個,經卡方測驗(χ2=1.57,=0.20>0.05),符合1﹕3期望比,從F2:3家系的反應型看,純合抗病家系數有67個,純合感病家系數有77個,抗感分離家系數150個,經卡方測驗(χ2=1.71,=0.42>0.05),符合1﹕2﹕1期望比(表1),說明小麥抗病材料Z501的抗性由1對主效基因控制。
2.3 BSA與基因定位
根據BSA結果,在抗、感池之間共有2 057個差異SNP位點,依據小麥660K SNP的染色體參考位置,其中,2B染色體上有904個差異SNP,剩余的差異SNP分布于其他染色體上,而且在2B染色體上抗感池差異性位點與親本間差異性位點重疊比例最高(圖1-d)。按照每1 Mb統計SNP差異位點數目,發現2B上的差異SNP大部分在500—600 Mb(染色體總長度為800 Mb)區間內(圖1-e)。為了確定目標基因所在區間,在區間500—600 Mb內開發了76個染色體特異性AQP標記,經過親本、抗感池以及小群體驗證,其中24個標記特異性良好,并在F2群體上進行基因分型,將標記檢測的基因型數據和表型數據進行整合,利用Joinmap軟件繪制出全長22.7 cM的遺傳圖譜,最終將目標基因鎖定在AQP標記和間隔1.1 cM區間上(圖1-f),對應物理區間為572.628—577.452 Mb(圖1-g),與全基因組關聯分析鎖定的區間范圍基本一致,對該位點暫命名為。
2.4 比較基因組分析
調取目標區域在中國春參考基因組中物理區間(575.706—576.587 Mb)內所有高可信度基因,分別與10+Genome等不同六倍體小麥品種基因組以及四倍體基因組進行比對分析。目標區域在中國春小麥參考基因組上跨度為0.26 Mb,對應的其他六倍體基因組區間在0.26—0.65 Mb不等,在六倍體斯卑爾脫小麥、四倍體硬粒小麥、野生二粒小麥和擬斯卑爾脫山羊草跨度在0.27—0.66 Mb不等(圖3),說明目標區域在從二倍體到六倍體小麥演化過程中發生了片段的插入缺失。進一步分析發現,該區間的6個高可信度小麥基因,基本都能在其他小麥材料中找到同源基因(圖3),且基因排列順序相同,極個別基因組存在某個基因的丟失和插入現象,但整體共線性良好,可以利用參考基因組信息進行候選基因預測分析。
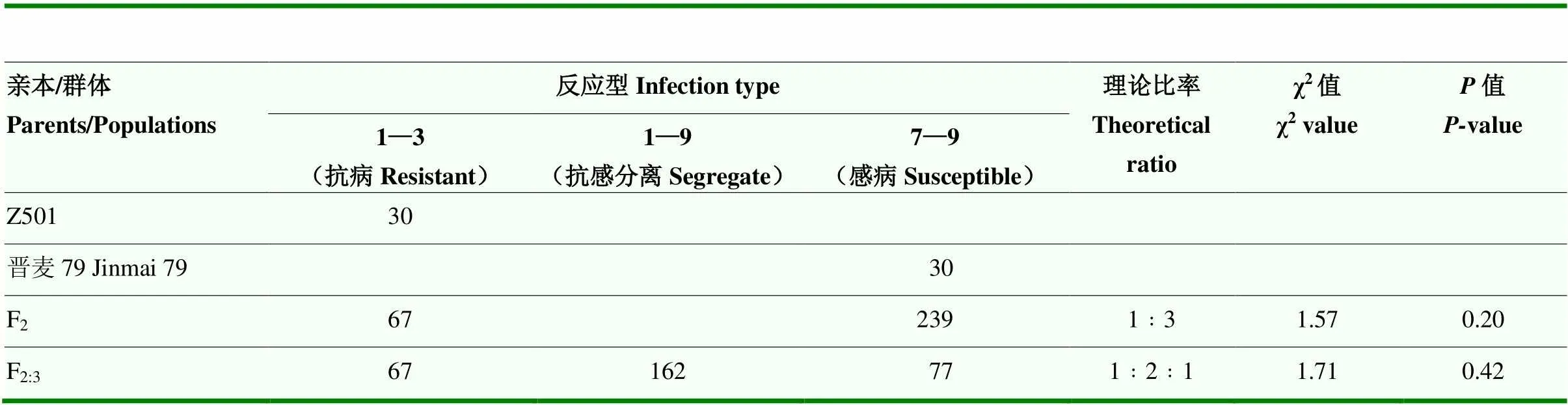
表1 晉麥79×Z501群體田間抗性反應統計及遺傳分析

圖2 小麥材料Z501和晉麥79田間抗病表現
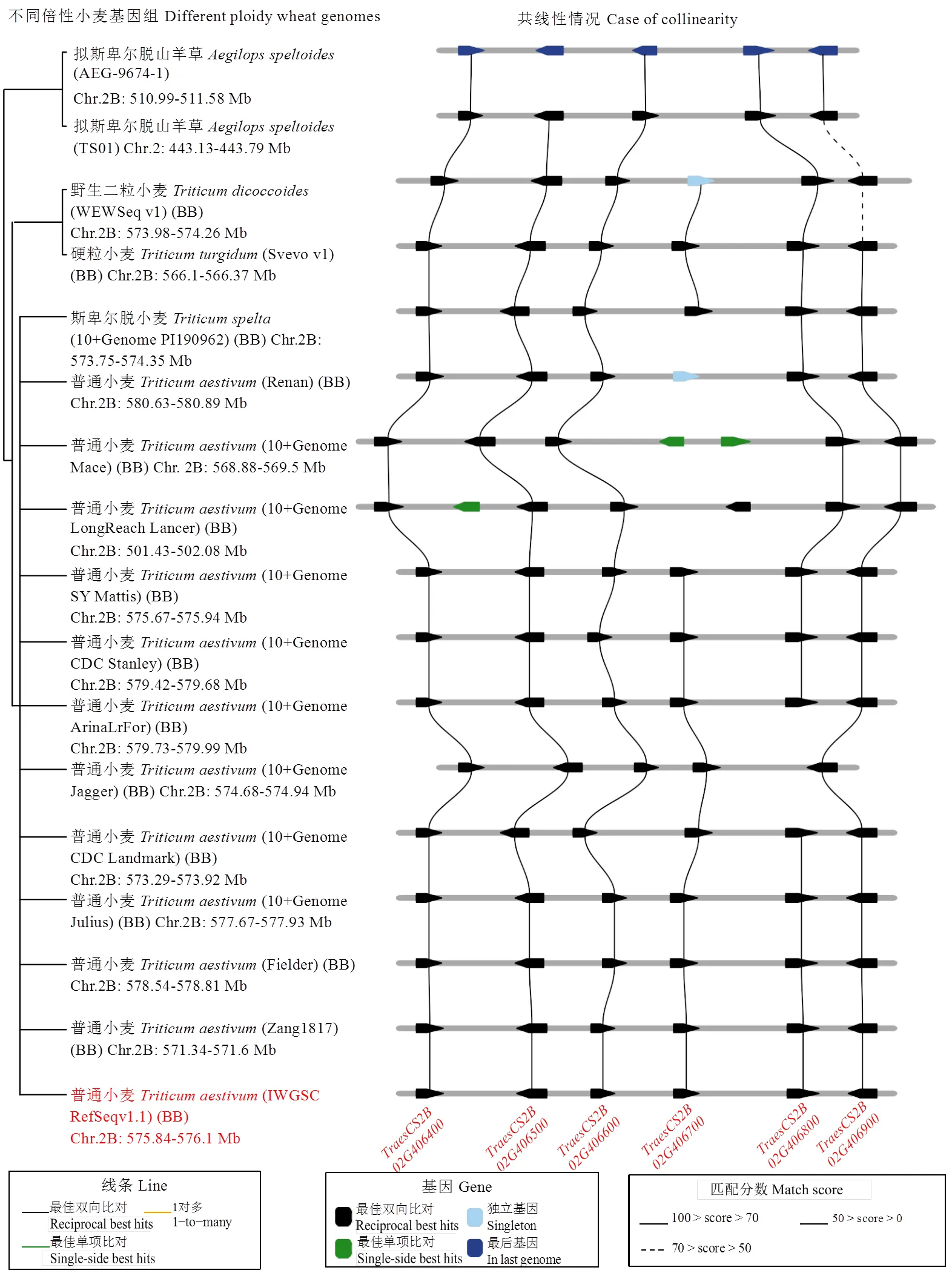
圖3 YrZ501物理區間及其與不同倍性小麥材料基因組的共線性分析
2.5 候選基因預測
根據中國春參考基因組的注釋信息,在的定位區間內共含有12個基因(圖4-a),根據Z501和晉麥79成株期接種條銹菌后的6個時間點(0、1、2、4、7和14 d)轉錄表達量分析(圖4-b),6個低可信基因均未表達,也沒有表達,和在接種前后表達量差異不顯著,、和在接種前后的表達量差異顯著,且抗病Z501和感病晉麥79之間也存在差異。其中,編碼GATA轉錄因子;屬于的一種可變剪切,編碼SH3P2蛋白;、分別是的2種可變剪切,均編碼B-box鋅指結構蛋白。根據前人的研究,三者均與抗病有關,因此,推斷、和可能是的候選基因。
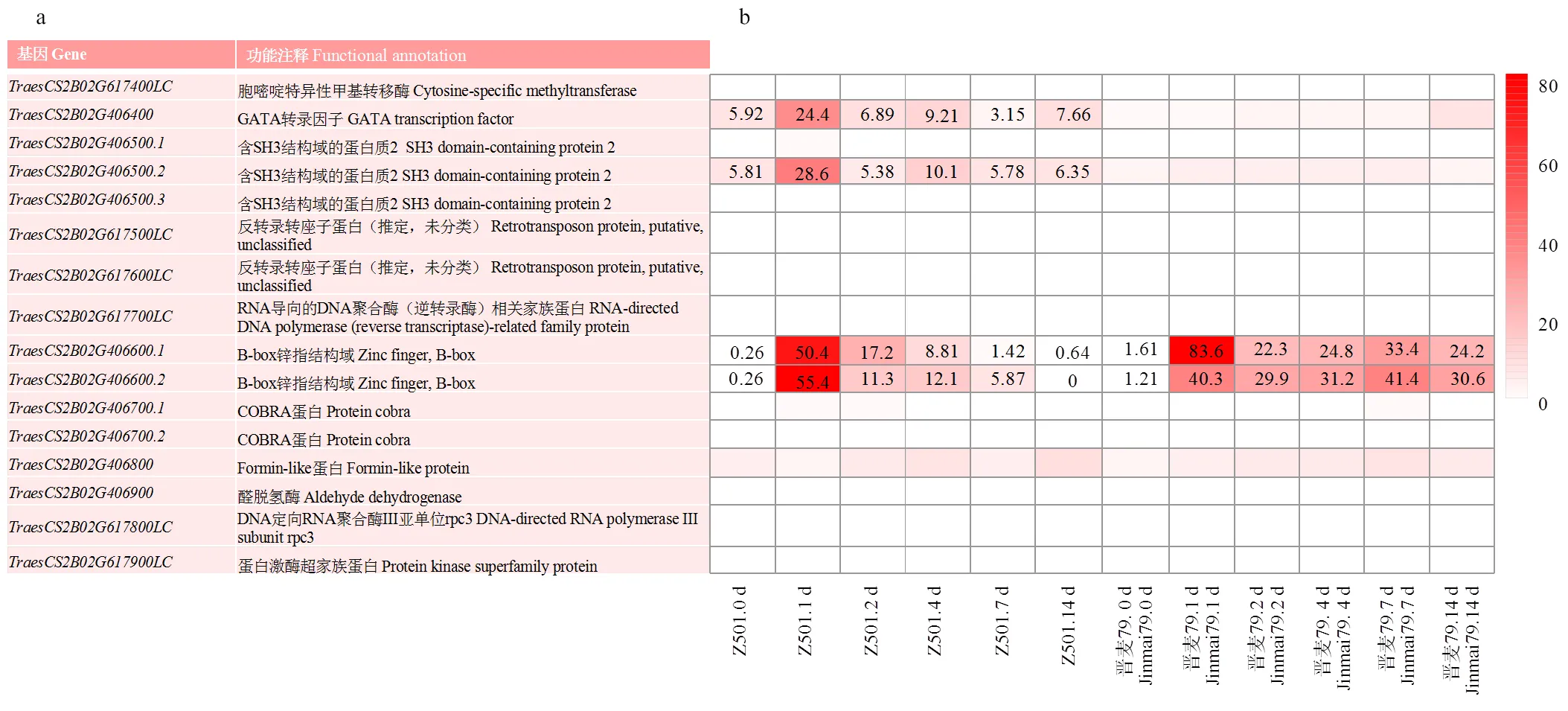
a:候選區間內基因功能注釋信息;b:親本Z501與晉麥79的轉錄組表達量
2.6 候選基因關聯分析
針對2.5中初步選定的3個候選基因,調取其在455份小麥重測序的序列變異,并進行候選基因關聯分析,取-log10()=3閾值,結果顯示,基因和中均未發現顯著變異SNP位點(圖5-a—b),僅在中有一個顯著性變異SNP位點(G1369A)(圖5-c),抗病親本Z501在該位點處與中國春參考基因組相同,均為G,而感病親本晉麥79在該位點處發生了變異,由G變為A,根據1.8中差異SNP分類原則,該位點(G1369A)在2個可變剪切的轉錄本中均未引起氨基酸編碼變化(TCG和TCA均編碼絲氨酸)(圖5-d),但可能與可變剪切有關,同時,該位點(G1369A)的不同單倍型表型之間也存在極顯著差異(圖5-e)。進一步分析發現,在G1369A位點下游還有2個引起氨基酸改變的變異位點——G1377A和G1431A,分別引起纈氨酸(GTT)到異亮氨酸(ATT)和纈氨酸(GTG)到甲硫氨酸(ATG)的改變(圖5-d),但2個位點在455份重測序材料中所占比例只有0.87%,屬于稀有變異,在利用GAPIT分析時被過濾掉,因此,未進行顯著性檢驗。綜上,推測的可變剪切轉錄本為的重要抗病候選基因。
3 討論
3.1 關聯分析與連鎖作圖相結合可有效解析復雜性狀
近年來,關聯分析和連鎖作圖相結合來解析復雜數量性狀已成為越來越重要的方法。關聯分析以數百個種質組成的自然群體為研究材料,由于歷史重組事件的積累而產生的大量遺傳變異,可以顯著地增加變異范圍,而且可以同時檢測同一基因座上的多個等位基因[38]。然而,關聯分析存在2個缺陷,一是關聯分析無法檢測稀有等位變異;二是當自然群體存在較為復雜的群體結構時,可能導致基因多態性位點與性狀的相關性并非由功能性等位基因引起,從而出現假陽性結果[38]。與之相比,連鎖作圖已被證明可以有效地剖析并定位復雜性狀的基因/QTL,但由于所應用的親本數較少,后代群體重組的機會有限,因此,當等位基因在雙親中無差異時,基因/QTL無法通過連鎖作圖進一步實現精細定位[39]。不論是連鎖作圖,還是關聯分析在進行QTL檢測時均具有一定的優缺點,所以充分利用2種方法的優勢,將2種方法相結合不但能夠顯著提高基因/QTL的檢測效率,還能有助于發現可靠、穩定的基因/QTL。例如Zhang等[40]利用228個重組自交系定位到與黃曲霉毒素積累顯著相關的QTL(),隨后又用437份自交系和558 629個SNP標記對黃曲霉毒素進行全基因組關聯分析,鑒定出了25個顯著的SNP位點,且這些SNP位點大部分與共定位,最終將縮小到1.5 kb區間內;常立國等[41]利用150份重組自交系群體和139份自然材料組成的關聯群體同時對持綠相關性狀進行定位,共同檢測到4個遺傳穩定的共定位遺傳區段;ZhaNG等[42]利用375份油菜自然群體和150個DH遺傳群體,通過關聯和連鎖分析共定位到一個新的控制油酸含量的主效QTL,并將其精細定位在76 kb的區間;劉凱等[43]利用小麥重組自交系群體和由205個品種(系)構成的自然群體,對兩群體的莖稈斷裂強度相關性狀進行連鎖作圖和全基因組關聯分析。在4B染色體上,檢測到9個與小麥莖稈斷裂強度相關QTL,最后定位到6.7 cM區間內。課題組前期利用411份CIMMYT和ICARDA春小麥材料通過關聯分析在2BL染色體上定位出大量的主效穩定的基因/QTL,并利用遺傳群體分別確認了和的可靠性。本試驗通過晉麥79×Z501遺傳群體的連鎖作圖驗證了另外一個GWAS主效位點,為快速發掘有效抗條銹病基因/QTL資源提供了有效手段。
3.2 多組學研究手段結合新方法是實現抗病基因快速克隆的有效途徑
隨著測序技術不斷更新迭代以及成本的下降,海量數據呈井噴式涌現出來。使得科研工作者可以采用大數據分析、多組學研究手段以及多種新方法結合分析,迅速克隆目的基因并對其調控機理進行全面解析。如,利用3 000多份水稻重測序數據,結合全基因組關聯分析快速挖掘了水稻廣譜抗稻瘟病遺傳位點[44];整合分析番茄轉錄組和代謝組,迅速鎖定了法卡林二醇的生物合成基因簇參與番茄對病害脅迫的反應[45];利用RNA深度測序發現miRNA參與植物的抗病性,并揭示了多個miRNA參與病害調控的機理[46];采用結構生物學的手段解析了抗病小體結構,揭示了NLR蛋白調控免疫的新機理等[47]。近些年,小麥在多組學上也取得了一系列突破和進展,截至目前,二倍體、四倍體、六倍體小麥的基因組精細圖譜相繼完成[48],其他六倍體小麥品種基因組測序和組裝,如國外10+Pan-genome[49],中國自主測定組裝科農9204等也陸續釋放[50],此外,小麥的大規模重測序、外顯子測序、全生育期轉錄組、代謝組工作也如火如荼開展起來[19, 21, 23, 51-53]。這些海量的基因組數據提供了豐富的參考信息和技術手段,極大促進了對小麥抗病基因挖掘。例如,突變體R基因富集測序(mutant R gene enrichment sequencing,MutRenSeq)、突變體目標染色體測序(mutant chromosome sequencing,MutChromSeq)、目標染色體長片段組裝(targeted chromosome-based cloning via long-range assembly,TACCA)等技術體系的提出,與傳統的圖位克隆方法相比,無需構建大規模的遺傳作圖群體,無需構建BAC文庫,不完全依賴參考基因組,對基因的克隆快速、高效、準確[54];近年來衍生出來的利用關聯分析結合R基因富集測序的方法(AgRenSeq),可以快速同時實現對多個抗稈銹病基因的克隆[14];利用高密度SNP芯片、重測序等基因組大數據通過關聯分析和連鎖作圖,并利用轉錄組、變異組等信息基于每個核苷酸多態性的功能重要性進行評估,快速開展了小麥成株期抗條銹病候選基因的克隆[10]和抗莖基腐病候選基因的克隆[55]等工作。充分利用當前小麥基因組學及功能基因組學最新的研究成果,整合現有的技術和方法,相互補充和完善,是發掘和克隆重要基因的一條有效途徑。本研究同樣整合了多組學手段,綜合分析快速發掘出的候選基因,為進一步克隆并驗證其功能奠定了堅實基礎。
3.3 YrZ501與2BL染色體上其他抗病位點的關系及潛在的育種價值
來源于春小麥Z501,被定位在IWGSC RefSeq v1.1參考基因組2BL染色體的575.706—576.587 Mb約119 kb范圍。根據前人對于小麥抗病QTL元分析的結果[1, 56],在2BL染色體區段的310—783 Mb(幾乎整個染色體長臂)都有大量的抗病QTL定位出來,其中,包含已正式命名的基因、/、、和,這些基因都認為是全生育期抗性基因,目前,除了和外,其他基因對中國當前的條銹菌流行小種基本失效[1],且和分別來自斯卑爾脫小麥(ssp.var.)和硬粒小麥(),因此,從抗性類型、來源等推斷,與二者不同。前人在該染色體區段已定位出多個成株期抗病QTL,包括CIMMYT春小麥的、美國冬小麥Druchamp的-、德國春小麥Naxos的-、意大利冬小麥Aquileja的-、法國冬小麥Camp Remy的-、中國冬小麥陜麥155的、中國冬小麥秦農142的-等[1, 56]。與以上部分位點有重疊,很可能為同一個抗性位點,但需要進一步的等位性測試。除了和-,其他位點基本都處于粗定位階段,存在定位精確度低、區間大等問題,極大限制了其在小麥抗病育種中的應用。本研究針對候選區間的差異SNP開發了相應的AQP標記,可用于輔助選擇,為下一步小麥抗銹病分子育種應用提供了標記資源。
4 結論
結合關聯分析和連鎖作圖在小麥2BL染色體上共定位到一個抗條銹病主效基因,并通過基因功能注釋、轉錄組數據分析、比較基因組分析、候選基因關聯分析預測為重要候選基因。
[1] 韓德俊, 康振生. 中國小麥品種抗條銹病現狀及存在問題與對策. 植物保護, 2018, 44(5): 1-12.
HAN D J, KANG Z S. Current status and future strategy in breeding wheat for resistance to stripe rust in China. Plant Protection, 2018, 44(5): 1-12. (in Chinese)
[2] 康振生, 王曉杰, 趙杰, 湯春蕾, 黃麗麗. 小麥條銹菌致病性及其變異研究進展. 中國農業科學, 2015, 48(17): 3439-3453.
KANG Z S, WANG X J, ZHAO J, TANG C L, HUANG L L. Advances in research of progress evariation of the wheat stripe rust fungusf. sp.. Scientia Agricultura Sinica, 2015, 48(17): 3439-3453. (in Chinese)
[3] 鄧一文, 劉裕強, 王靜, 陳學偉, 何祖華. 農作物抗病蟲研究的戰略思考. 中國科學: 生命科學, 2021, 51(10): 1435-1446.
DENG Y W, LIU Y Q, WANG J, CHEN X W, HE Z H. Strategic thinking and research on crop diseases and pest resistance in China. Scientia Sinica (Vitae), 2021, 51(10): 1435-1446. (in Chinese)
[4] KOURELIS J, van der HOORN R A L. Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. The Plant Cell, 2018, 30(2): 285-299.
[5] KRATTINGER S G, LAGUDAH E S, SPIELMEYER W, SINGH R P, HUERTA-ESPINO J, MCFADDEN H, BOSSOLINI E, SELTER L L, KELLER B. A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science, 2009, 323(5919): 1360-1363.
[6] KRATTINGER S G, KANG J, BR?UNLICH S, BONI R, CHAUHAN H, SELTER L L, ROBINSON M D, SCHMID M W, WIEDERHOLD E, HENSEL G, KUMLEHN J, SUCHER J, MARTINOIA E, KELLER B. Abscisic acid is a substrate of the ABC transporter encoded by the durable wheat disease resistance gene Lr34. New Phytologist, 2019, 223(2): 853-866.
[7] FU D L, UAUY C, DISTELFELD A, BLECHL A, EPSTEIN L, CHEN X M, SELA H N, FAHIMA T, DUBCOVSKY J. A kinase-START gene confers temperature-dependent resistance to wheat stripe rust. Science, 2009, 323(5919): 1357-1360.
[8] GOU J Y, LI K, WU K T, WANG X D, LIN H Q, CANTU D, UAUY C, DOBON-ALONSO A, MIDORIKAWA T, INOUE K, SáNCHEZ J, FU D L, BLECHL A, WALLINGTON E, FAHIMA T, MEETA M, EPSTEIN L, DUBCOVSKY J. Wheat stripe rust resistance protein WKS1 reduces the ability of the thylakoid-associated ascorbate peroxidase to detoxify reactive oxygen species. The Plant Cell, 2015, 27(6): 1755-1770.
[9] MOORE J W, HERRERA-FOESSEL S, LAN C X, SCHNIPPENKOETTER W, AYLIFFE M, HUERTA-ESPINO J, LILLEMO M, VICCARS L, MILNE R, PERIYANNAN S, KONG X Y, SPIELMEYER W, TALBOT M, BARIANA H, PATRICK J W, DODDS P, SINGH R, LAGUDAH E. A recently evolved hexose transporter variant confers resistance to multiple pathogens in wheat. Nature Genetics, 2015, 47(12): 1494-1498.
[10] WU J H, YU R, WANG H Y, ZHOU C E, HUANG S, JIAO H X, YU S Z, NIE X J, WANG Q L, LIU S J, SONG W N, SINGH R P, BHAVANI S, KANG Z S, HAN D J, ZENG Q D. A large-scale genomic association analysis identifies the candidate causal genes conferring stripe rust resistance under multiple field environments. Plant Biotechnology Journal, 2021, 19(1): 177-191.
[11] BEVAN M W, UAUY C, WULFF B B H, ZHOU J, KRASILEVA K, CLARK M D. Genomic innovation for crop improvement. Nature, 2017, 543(7645): 346-354.
[12] BETTGENHAEUSER J, KRATTINGER S G. Rapid gene cloning in cereals. Theoretical and Applied Genetics,2019, 132(3): 699-711.
[13] YANO K, YAMAMOTO E, AYA K, TAKEUCHI H, LO P C, HU L, YAMASAKI M, YOSHIDA S, KITANO H, HIRANO K, MATSUOKA M. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nature Genetics, 2016, 48(8): 927-934.
[14] ARORA S, STEUERNAGEL B, GAURAV K, CHANDRAMOHAN S, LONG Y M, MATNY O, JOHNSON R, ENK J, PERIYANNAN S, SINGH N, ASYRAF MD HATTA M, ATHIYANNAN N, CHEEMA J, YU G T, KANGARA N, GHOSH S, SZABO L J, POLAND J, BARIANA H, JONES J D G, BENTLEY A R, AYLIFFE M, OLSON E, XU S S, STEFFENSON B J, LAGUDAH E, WULFF B B H. Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nature Biotechnology, 2019, 37(2): 139-143.
[15] LIU F, ZHAO Y S, BEIER S, JIANG Y, THORWARTH P, LONGIN C F H, GANAL M, HIMMELBACH A, REIF J C, SCHULTHESS A W. Exome association analysis sheds light onto leaf rust () resistance genes currently used in wheat breeding (L.). Plant Biotechnology Journal, 2020, 18(6): 1396-1408.
[16] GUO Z F, CHEN D J, ALQUDAH A M, R?DER M S, GANAL M W, SCHNURBUSCH T. Genome-wide association analyses of 54 traits identified multiple loci for the determination of floret fertility in wheat. The New Phytologist, 2017, 214(1): 257-270.
[17] LI L, MAO X G, WANG J Y, CHANG X P, REYNOLDS M, JING R L. Genetic dissection of drought and heat-responsive agronomic traits in wheat. Plant, Cell & Environment, 2019, 42(9): 2540-2553.
[18] SUN C W, DONG Z D, ZHAO L, REN Y, ZHANG N, CHEN F. The Wheat 660K SNP array demonstrates great potential for marker- assisted selection in polyploid wheat. Plant Biotechnology Journal, 2020, 18(6): 1354-1360.
[19] CHEN J, HU X, SHI T T, YIN H R, SUN D F, HAO Y F, XIA X C, LUO J, FERNIE A R, HE Z H, CHEN W. Metabolite-based genome-wide association study enables dissection of the flavonoid decoration pathway of wheat kernels. Plant Biotechnology Journal, 2020, 18(8): 1722-1735.
[20] ZHOU C E, LIU D, ZHANG X, WU Q M, LIU S J, ZENG Q D, WANG Q L, WANG C F, LI C L, SINGH R P, BHAVANI S, KANG Z S, HAN D J, ZHENG W J, WU J H. Combined linkage and association mapping reveals two major QTL for stripe rust adult plant resistance in Shaanmai 155 and their haplotype variation in common wheat germplasm. The Crop Journal, 2022, 10(3): 783-792.
[21] WANG T T, SU N, LU J N, ZHANG R P, SUN X M, SONG W N. Genome-wide association studies of peduncle length in wheat under rain-fed and irrigating field conditions. Journal of Plant Physiology, 2023, 280: 153854.
[22] GUO W L, XIN M M, WANG Z H, YAO Y Y, HU Z R, SONG W J, YU K H, CHEN Y M, WANG X B, GUAN P F, APPELS R, PENG H R, NI Z F, SUN Q X. Origin and adaptation to high altitude of Tibetan semi-wild wheat. Nature Communications, 2020, 11: 5085.
[23] ZHOU Y, ZHAO X B, LI Y W, XU J, BI A Y, KANG L P, XU D X, CHEN H F, WANG Y, WANG Y G, LIU S Y, JIAO C Z,
LU H F, WANG J, YIN C B, JIAO Y L, LU F. Triticum population sequencing provides insights into wheat adaptation. Nature Genetics, 2020, 52(12): 1412-1422.
[24] HAO C Y, JIAO C Z, HOU J, LI T, LIU H X, WANG Y Q, ZHENG J, LIU H, BI Z H, XU F F, ZHAO J, MA L, WANG Y M, MAJEED U, LIU X, APPELS R, MACCAFERRI M, TUBEROSA R, LU H F, ZHANG X Y. Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China. Molecular plant, 2020, 13(12): 1733-1751.
[25] LI M X, YEUNG J M Y, CHERNY S S, SHAM P C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Human Genetics, 2012, 131(5): 747-756.
[26] TEAM R. R: a language and environment for statistical computing. Computer Science, 2014.
[27] YANG J, LEE S H, GODDARD M E, VISSCHER P M. GCTA: a tool for genome-wide complex trait analysis. The American Journal of Human Genetics, 2011, 88(1): 76-82.
[28] LINE R, QAYOUM A. Virulence, aggressiveness, evolution, and distribution of races of(the cause of stripe rust of wheat) in North America 1968-87. US Department of Agriculture Technical Bulletin, 1992, 74, 1788.
[29] CHEN X M. Pathogens which threaten food security: Puccinia striiformis, the wheat stripe rust pathogen. Food e, 2020, 12(2): 239-251.
[30] LIU S J, WANG X T, ZHANG Y Y, JIN Y G, XIA Z H, XIANG M J, HUANG S, QIAO L Y, ZHENG W J, ZENG Q D, WANG Q L, YU R, SINGH R P, BHAVANI S, KANG Z S, HAN D J, WANG C F, WU J H. Enhanced stripe rust resistance obtained by combining Yr30 with a widely dispersed, consistent QTL on chromosome arm 4BL. Theoretical and e Genetics, 2022, 135(1): 351-365.
[31] Van OOIJEN J W. JoinMap4, software for the calculation of genetic linkage maps in experimental populations. Wageningen, The Netherlands, Kyazma BV, 2006.
[32] KOSAMBI D D. The estimation of map distances from recombination values. Annals of Eugenics, 1943, 12(1): 172-175.
[33] VOORRIPS R E. MapChart: Software for the graphical presentation of linkage maps and QTLs. Journal of Heredity, 2002, 93(1): 77-78.
[34] APPELS R, EVERSOLE K, STEIN N, FEUILLET C, KELLER B, ROGERS J, POZNIAK C, CHOULET F, DISTELFELD A, POLAND J, RONEN G, SHARPE A, BARAD O, BARUCH K, KEEBLE-GAGNèRE G, MASCHER M, BEN-ZVI G, JOSSELIN A, HIMMELBACH A, BALFOURIER F, GUTIERREZ-GONZALEZ J J, HAYDEN M, KOH C, MUEHLBAUER G, PASAM R, PAUX E, RIGAULT P, TIBBITS J, TIWARI V, SPANNAGL M, LANG D, GUNDLACH H, HABERER G, MAYER K, ORMANBEKOVA D, PRADE V M, ?IMKOVá H, WICKER T, SWARBRECK D, RIMBERT H, FELDER M, GUILHOT N, KAITHAKOTTIL G G, KEILWAGEN J, LEROY P, LUX T M, TWARDZIOK S, VENTURINI L, JUHáSZ A, ABROUK M, FISCHER I, UAUY C, BORRILL P, RAMíREZ-GONZáLEZ R, ARNAUD D, CHALABI S, CHALHOUB B, CORY A, DATLA R, DAVEY M, JACOBS J, ROBINSON S J, STEUERNAGEL B, VAN EX F, WULFF B, BENHAMED M, BENDAHMANE A, CONCIA L, LATRASSE D, BARTO? J, BELLEC A, BERGèS H, DOLE?EL J, FRENKEL Z, GILL B, KOROL A, LETELLIER T, OLSEN O, SINGH K, VALáRIK M, VAN DER VOSSEN E V D, VAUTRIN S, WEININGS, FAHIMA T, GLIKSON V, RAATS D, ?íHALíKOVá J, TOEGELOVá H, VRáNA J, SOURDILLE P, DARRIER B, BARABASCHI D, CATTIVELLI L, HERNáNDEZ P, GáLVEZ S, BUDAK H, JONES J D G, WITEK K, YU G T, SMALL I, MELONEK J, ZHOU R N, BELOVA T, KANYUKA K, KING R, NILSEN K, WALKOWIAK S, CUTHBERT R, KNOX R, WIEBE K, XIANG D, ROHDE A, GOLDS T, ?í?KOVá J, AKP?NAR B A, BIYIKLIOGLU S, GAO L L, N'DAIYE A, KUBALAU0301KOVAU0301 M, ?AFá? J, ALFAMA F, ADAM-BLONDON A, FLORES R, GUERCHE C, LOAEC M, QUESNEVILLE H, CONDIE J, ENS J, MACLACHLAN R, TAN Y F, ALBERTI A, AURY J, BARBE V, COULOUX A, CRUAUD C, LABADIE K, MANGENOT S, WINCKER P, KAUR G, LUO M, SEHGAL S, CHHUNEJA P, GUPTA O, JINDAL S, KAUR P, MALIK P, SHARMA P, YADAV B, SINGH N, KHURANA J, CHAUDHARY C, KHURANA P, KUMAR V, MAHATO A K, MATHUR S, SEVANTHI A, SHARMA N, TOMAR R S S, HOLU?OVá K, PLíHAL O, CLARK M, HEAVENS D, KETTLEBOROUGH G, WRIGHT J, BALCáRKOVá B, HU Y Q, SALINA E, RAVIN N, SKRYABIN K, BELETSKY A, KADNIKOV V, MARDANOV A, NESTEROV M, RAKITIN A, SERGEEVA E, HANDA H, KANAMORI H, KATAGIRI S, KOBAYASHI F, NASUDA S, TANAKA T, WU J, CATTONARO F, MIN J M, KUGLER K G, PFEIFER M, SANDVE S, XUN X, ZHAN B, BATLEY J, BAYER P, EDWARDS D, HAYASHI S, TULPOVá Z, VISENDI P, CUI L C, DU X H, FENG K W, NIE X J, TONG W, WANG L. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science, 2018, 361(6403): eaar7191.
[35] CHEN Y M, SONG W J, XIE X M, WANG Z H, GUAN P F, PENG H R, JIAO Y N, NI Z F, SUN Q X, GUO W L. A collinearity- incorporating homology inference strategy for connecting emerging assemblies in thetribe as a pilot practice in the plant pangenomic era. Molecular plant, 2020, 13(12): 1694-1708.
[36] MA S W, WANG M, WU J H, GUO W L, CHEN Y M, LI G W, WANG Y P, SHI W M, XIA G M, FU D L, KANG Z S, NI F. WheatOmics: A platform combining multiple omics data to accelerate functional genomics studies in wheat. Molecular plant, 2021, 14(12): 1965-1968.
[37] WANG J B, ZHANG Z W. GAPIT version 3: boosting power and accuracy for genomic association and prediction. Genomics, Proteomics & Bioinformatics, 2021, 19(4): 629-640.
[38] YU J M, PRESSOIR G, BRIGGS W H, BI I V, YAMASAKI M, DOEBLEY J F, MCMULLEN M D, GAUT B S, NIELSEN D M, HOLLAND J B, KRESOVICH S, BUCKLER E S. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics, 2006, 38(2): 203-208.
[39] LU Y L, ZHANG S H, SHAH T, XIE C X, HAO Z F, LI X H, FARKHARI M, RIBAUT J M, CAO M J, RONG T Z, XU Y B. Joint linkage-linkage disequilibrium mapping is a powerful approach to detecting quantitative trait loci underlying drought tolerance in maize. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(45): 19585-19590.
[40] ZHANG Y, CUI M, ZHANG J M, ZHANG L, LI C L, KAN X, SUN Q, DENG D X, YIN Z T. Confirmation and fine mapping of a major QTL for aflatoxin resistance in maize using a combination of linkage and association mapping. Toxins, 2016, 8(9): 258.
[41] 常立國, 何坤輝, 劉建超. 多環境下玉米保綠相關性狀遺傳位點的挖掘. 中國農業科學, 2022, 55(16): 3071-3081.
CHANG L G, HE K H, LIU J C. Mining of genetic locus of maize stay-green related traits under multi-environments. Scientia Agricultura Sinica, 2022, 55(16): 3071-3081. (in Chinese)
[42] ZHANG X X, GUAN Z R, LI Z L, LIU P, MA L L, ZHANG Y C, PAN L, HE S J, ZHANG Y L, LI P, GE F, ZOU C Y, HE Y C, GAO S B, PAN G T, SHEN Y O. A combination of linkage mapping and GWAS brings new elements on the genetic basis of yield-related traits in maize across multiple environments. Theoretical and Applied Genetics,2020, 133(10): 2881-2895.
[43] 劉凱, 鄧志英, 張瑩, 王芳芳, 劉佟佟, 李青芳, 邵文, 趙賓, 田紀春, 陳建省. 小麥莖稈斷裂強度相關性狀QTL的連鎖和關聯分析. 作物學報, 2017, 43(4): 483-495.
LIU K, DENG Z Y, ZHANG Y, WANG F F, LIU T T, LI Q F, SHAO W, ZHAO B, TIAN J C, CHEN J S. Linkage analysis and genome-wide association study of QTLs controlling stem-breaking- strength-related traits in wheat. Acta Agronomica Sinica, 2017, 43(4): 483-495. (in Chinese)
[44] LI W T, ZHU Z W, CHERN M, YIN J J, YANG C, RAN L, CHENG M P, HE M, WANG K, WANG J, ZHOU X G, ZHU X B, CHEN Z X, WANG J C, ZHAO W, MA B T, QIN P, CHEN W L, WANG Y P, LIU J L, WANG W M, WU X J, LI P, WANG J R, ZHU L H, LI S G, CHEN X W. A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell, 2017, 170(1): 114-126.e15.
[45] JEON J E, KIM J G, FISCHER C R, MEHTA N, DUFOUR- SCHROIF C, WEMMER K, MUDGETT M B, SATTELY E. A pathogen-responsive gene cluster for highly modified fatty acids in tomato. Cell, 2020, 180(1): 176-187.e19.
[46] YANG L, HUANG H. Roles of small RNAs in plant disease resistance. Journal of Integrative Plant Biology, 2014, 56(10): 962-970.
[47] WANG J Z, HU M J, WANG J, QI J F, HAN Z F, WANG G X, QI Y J, WANG H W, ZHOU J M, CHAI J J. Reconstitution and structure of a plant NLR resistosome conferring immunity. Science, 2019, 364(6435): eaav5870.
[48] 葸瑋, 郝晨陽, 李甜, 劉云川, 焦成智, 王化俊, 張學勇. 基因組時代-麥類基因組學研究現狀及趨勢. 植物遺傳資源學報, 2022, 23(4): 929-942.
XI W, HAO C Y, LI T, LIU Y C, JIAO C Z, WANG H J, ZHANG X Y. The ear genomics: current status and future trend of genomics research triticeae crops. Journal of Plant Genetic Resources, 2022, 23(4): 929-942. (in Chinese)
[49] WALKOWIAK S, GAO L L, MONAT C, HABERER G, KASSA M T, BRINTON J, RAMIREZ-GONZALEZ R H, KOLODZIEJ M C, DELOREAN E, THAMBUGALA D, KLYMIUK V, BYRNS B, GUNDLACH H, BANDI V, SIRI J N, NILSEN K, AQUINO C, HIMMELBACH A, COPETTI D, BAN T, VENTURINI L, BEVAN M, CLAVIJO B, KOO D H, ENS J, WIEBE K, N’DIAYE A, FRITZ A K, GUTWIN C, FIEBIG A, FOSKER C, FU B X, ACCINELLI G G, GARDNER K A, FRADGLEY N, GUTIERREZ-GONZALEZ J, HALSTEAD-NUSSLOCH G, HATAKEYAMA M, KOH C S, DEEK J, COSTAMAGNA A C, FOBERT P, HEAVENS D, KANAMORI H, KAWAURA K, KOBAYASHI F, KRASILEVA K, KUO T, MCKENZIE N, MURATA K, NABEKA Y, PAAPE T, PADMARASU S, PERCIVAL-ALWYN L, KAGALE S, SCHOLZ U, JUN S S, JULIANA P, SINGH R, SHIMIZU-INATSUGI R, SWARBRECK D, COCKRAM J, BUDAK H, TAMESHIGE T, TANAKA T, TSUJI H, WRIGHT J, WU J Z, STEUERNAGEL B, SMALL I, CLOUTIER S, KEEBLE-GAGNèRE G, MUEHLBAUER G, TIBBETS J, NASUDA S, MELONEK J, HUCL P J, SHARPE A G, CLARK M, LEGG E, BHARTI A, LANGRIDGE P, HALL A, UAUY C, MASCHER M, KRATTINGER S G, HANDA H, SHIMIZU K K, DISTELFELD A, CHALMERS K, KELLER B, MAYER K F X, POLAND J, STEIN N, MCCARTNEY C A, SPANNAGL M, WICKER T, POZNIAK C J. Multiple wheat genomes reveal global variation in modern breeding. Nature, 2020, 588(7837): 277-283.
[50] SHI X L, CUI F, HAN X Y, HE Y L, ZHAO L, ZHANG N, ZHANG H, ZHU H D, LIU Z X, MA B, ZHENG S S, ZHANG W, LIU J J, FAN X L, SI Y Q, TIAN S Q, NIU J Q, WU H L, LIU X M, CHEN Z, MENG D Y, WANG X Y, SONG L Q, SUN L J, HAN J, ZHAO H, JI J, WANG Z G, HE X Y, LI R L, CHI X B, LIANG C Z, NIU B F, XIAO J, LI J M, LING H Q. Comparative genomic and transcriptomic analyses uncover the molecular basis of high nitrogen-use efficiency in the wheat cultivar Kenong 9204. Molecular Plant, 2022, 15(9): 1440-1456.
[51] CHENG H, LIU J, WEN J, NIE X J, XU L H, CHEN N B, LI Z X, WANG Q L, ZHENG Z Q, LI M, CUI L C, LIU Z H, BIAN J X, WANG Z H, XU S B, YANG Q, APPELS R, HAN D J, SONG W N, SUN Q X, JIANG Y. Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat. e Biology, 2019, 20(1): 136.
[52] LI A L, HAO C Y, WANG Z Y, GENG S F, JIA M L, WANG F, HAN X, KONG X C, YIN L J, TAO S, DENG Z Y, LIAO R Y, SUN G L, WANG K, YE X G, JIAO C Z, LU H F, ZHOU Y, LIU D C, FU X D, ZHANG X Y, MAO L. Wheat breeding history reveals synergistic selection of pleiotropic genomic sites for plant architecture and grain yield. Molecular Plant, 2022, 15(3): 504-519.
[53] RAMíREZ-GONZáLEZ R H, BORRILL P, LANG D, HARRINGTON S A, BRINTON J, VENTURINI L, DAVEY M, JACOBS J, VAN EX F, PASHA A, KHEDIKAR Y, ROBINSON S J, CORY A T, FLORIO T, CONCIA L, JUERY C, SCHOONBEEK H, STEUERNAGEL B, XIANG D, RIDOUT C J, CHALHOUB B, MAYER K X, BENHAMED M, LATRASSE D, BENDAHMANE A, CONSORTIUM I W G S, WULFF B H, APPELS R, TIWARI V, DATLA R, CHOULET F, POZNIAK C J, PROVART N J, SHARPE A G, PAUX E, SPANNAGL M, BR?UTIGAM A, UAUY C. The transcriptional landscape of polyploid wheat. e, 2018, 361(6403): eaar6089.
[54] KELLER B, WICKER T, KRATTINGER S G. Advances in wheat and pathogen genomics: Implications for disease control. Annual Review of Phytopathology, 2018, 56: 67-87.
[55] YANG X, ZHONG S B, ZHANG Q J, REN Y, SUN C W, CHEN F. A loss-of-function of the dirigent gene TaDIR-B1 improves resistance to Fusarium crown rot in wheat. Plant e Journal, 2021, 19(5): 866-868.
[56] CHEN X M, KANG Z S. Stripe Rust. Dordrecht: Springer Netherlands Press, 2017: 723.
Identification of Adult Plant Stripe Rust Resistance Candidate Genes ofby Gene Association and Transciptome Analysis in Wheat (L.)
1College of Agronomy, Northwest A&F University/State Key Laboratory of Crop Stress Biology for Arid Areas, Yangling 712100, Shaanxi;2College of Plant Protection, Northwest A&F University/State Key Laboratory of Crop Stress Biology for Arid Areas, Yangling 712100, Shaanxi
【Objective】Stripe rust, caused byf. sp.(), significantly reduced wheat production worldwide. Identification of stripe rust resistance genes is the foundation of improving wheat resistance breeding and revealing its genetic mechanism.【Method】A multi-omics approach combined with genome-wide association study (GWAS) was used for dissecting adult plant stripe rust resistance for wheat advanced breeding lines collected from International Maize and Wheat Improvement Center (CIMMYT) and International Centre for Agricultural Research in the Dry Areas (ICARDA) bread-wheat breeding programs. In the present study, a diversity panel of 411 wheat lines from CIMMYT and ICARDA was used for genome-wide association study and a major locus on chromosome arm 2BL was identified. In order to verify the stability of the locus, the resistant line Z501 with the resistance allele of the locus was crossed by the susceptible line Jinmai 79, and the locus tentatively namedwas successfully confirmed using linkage mapping based on F2:3genetic population of Jinmai 79×Z501. Then we performed candidate gene analysis based on gene annotation, comparative genome, transcriptome and gene-based association analysis. 【Result】Combining GWAS and linkage mapping results, thewas located in the physical interval of 0.26 Mb (575.706-576.587 Mb) on chromosome 2B. According to the annotation information of Chinese Spring reference genome IWGSC v1.1, there were six high confidence genes of 12 genes in this region. Using online website, the target interval in the Chinese spring reference genome was compared with other published different ploidy wheat genomes. The six high-confidence genes within this interval can basically be found homologous in other wheat lines, and the genes arranged in the same order, indicating that the interval may not have large fragment insertions, deletions and inversions. The above results showed that we can perform candidate gene prediction analysis based on the reference genome information. After analysis of their transcriptomic data between the resistant parent Z501 and susceptible parent Jimai 79, only three genes,,andshowed variable expression levels and were induced by stripe rust infection. Further, they encode GATA transcription factor, SH3 domain-containing protein 2 and zinc finger protein, respectively. Gene-based association analysis revealed that there was a significant SNP (G1369A) inthat was associated with stripe rust responses. Although this SNP (G1369A) did not cause amino acid coding changes (both TCG and TCA encode serine), it may be associated with alternative splicing. Moreover, it showed significant differences of the stripe rust responses between the different haplotypes (G1369A). Further analysis revealed two other variants G1377A and G1431A, that caused amino acid changes, i. e. valine (GTT) to isoleucine (ATT) and valine (GTG) to methionine (ATG), respectively. However, the two SNPs were rare variants as they accounting for only 0.87% of the 455 re-sequencing wheat accessions and they were not tested for significance. In summary,was preliminarily considered as an important candidate gene of. In addition, the corresponding AQP markers were developed based on the SNPs among thecandidate regions, which can be used to marker-assisted selection in molecular breeding application of wheat stripe rust resistance.【Conclusion】A candidate causal geneassociated with stripe rust resistance was successfully identified on wheat chromosome 2B using an integrated method of multi-omics and association analysis, which laid a solid foundation for further gene cloning and functional verification.
; stripe rust resistance;; integrative analysis of multiple omics; SH3P2
10.3864/j.issn.0578-1752.2023.08.001
2022-12-08;
2023-01-19
國家重點研發計劃(2021YFD1200600)、青海省重點研發與轉化計劃(2022-NK-125)
張旭,E-mail:zhxu@nwafu.edu.cn。韓金妤,E-mail:1021979347@qq.com。張旭和韓金妤為同等貢獻作者。通信作者韓德俊,E-mail:handj@nwsuaf.edu.cn。通信作者吳建輝,E-mail:wujianhui@nwsuaf.edu.cn
(責任編輯 李莉)
猜你喜歡
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:30
當代陜西(2021年17期)2021-11-06 03:21:36
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
當代陜西(2019年15期)2019-09-02 01:52:00
電子制作(2018年18期)2018-11-14 01:48:24
學苑創造·A版(2018年11期)2018-02-01 06:29:20
讀者(2017年5期)2017-02-15 18:04:18
山東工業技術(2016年15期)2016-12-01 05:31:22
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
