高效液相色譜法同時測定富馬酸伏諾拉生中活性成分含量及成鹽率研究*
2023-06-11 11:03:56王玲蘭龔翠云朱躍芳
中國藥業 2023年11期
王玲蘭,龔翠云,朱躍芳,朱 婧,陳 莉,宿 亮
(1. 株洲千金藥業股份有限公司,湖南 株洲 412000; 2. 湖南省株洲市食品藥品檢驗所,湖南 株洲 412000)
富馬酸伏諾拉生曾用名富馬酸沃諾拉贊,化學名為1-[5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1H - 吡咯- 3 - 基]-N- 甲基甲胺富馬酸鹽,是一種新型鉀離子競爭性酸阻滯劑(P-CAB),主要用于治療反流性食管炎、胃潰瘍、十二指腸潰瘍、幽門螺桿菌的根除治療等酸相關疾病[1-4]。其抑酸效果迅速、有效、持續,能規避傳統質子泵抑制劑(PPIs)對酸不穩定且個體差異大等缺點[5-10]。富馬酸伏諾拉生由1 分子伏諾拉生與1 分子富馬酸結合而成[11-14],但鹽基過量會直接影響藥物的純度,游離活性成分過量將影響藥物在體內的吸收與分布[15-17]。故有必要建立監測藥物反離子含量的分析方法進行藥物成鹽率研究,以評價不同批次原料藥成鹽率的差異。目前,有關富馬酸伏諾拉生的報道均集中在有關物質方面[18-23],僅針對其活性成分及雜質進行了含量測定,尚未見富馬酸含量測定及成鹽率研究相關報道。本研究中建立了同時測定富馬酸伏諾拉生中富馬酸和伏諾拉生含量的高效液相色譜法,并對多批次原料藥的成鹽率進行評估,為其質量控制提供參考。現報道如下。
1 儀器與試藥
1.1 儀器
Waters e2695 型高效液相色譜儀(美國Waters 公司);MS105DU 型電子天平(精度為0.01 mg),DSC3 型差示掃描量熱儀,均購自梅特勒- 托利多公司;KQ -500DE 型數控超聲波清洗器(昆山市超聲儀器有限公司,功率為400 W,頻率為40 kHz)。
1.2 試藥
富馬酸伏諾拉生(千金湘江藥業股份有限公司,批號分別為20181101,20181102,20181103,20190101,20190102,20190103,20190401,20190402,20190403);伏諾拉生對照品(美國Quality Control Chemicals 公司,批號為20-AUG-17-40,含量為99.6%);富馬酸對照品(中國食品藥品檢定研究院,批號為111541 -201803,含量為99.9%);乙腈、甲醇(色譜純,美國Spectrum 公司);磷酸(色譜純,天津市科密歐化學試劑有限公司);磷酸氫二鈉(分析純,天津市恒興化學試劑制造有限公司);水為屈臣氏飲用純凈水。
2 方法與結果
2.1 色譜條件
色譜柱:Agilent 5 TC-C18(2)柱(250 mm×4.6 mm,5 μm);流動相:0.05 mol/ L 磷酸鹽緩沖液(pH 6.8)-乙腈- 甲醇(17∶7∶6,V/V/V);流速:1.0 mL/ min;檢測波長:230 nm;柱溫:30 ℃;進樣量:10μL。
2.2 溶液制備
伏諾拉生對照品溶液:避光操作。取伏諾拉生對照品100 mg,精密稱定,置50 mL 容量瓶中,加流動相適量,超聲20 min 使溶解,加流動相定容,搖勻,作為伏諾拉生對照品貯備液。精密量取貯備液5 mL,置50 mL 容量瓶中,加流動相溶解并定容,制成每1 mL約含200μg伏諾拉生的溶液,搖勻,即得。
富馬酸對照品溶液:避光操作。取富馬酸對照品17 mg,精密稱定,置50 mL 容量瓶中,加流動相適量,超聲20 min 使溶解,加流動相定容,搖勻,作為富馬酸對照品貯備液。精密量取貯備液10 mL,置50 mL 容量瓶中,加流動相溶解并定容,制成每1 mL 約含68μg 富馬酸的溶液,搖勻,即得。
供試品溶液:避光操作。取樣品27 mg,精密稱定,置100 mL 容量瓶中,加流動相適量,超聲20 min 使溶解,加流動相定容,搖勻,即得。
2.3 方法學考察
專屬性試驗:取空白溶劑(流動相)、伏諾拉生對照品溶液、富馬酸對照品溶液、供試品溶液各適量,按2.1項下色譜條件依次進樣測定。結果供試品溶液色譜中,富馬酸及伏諾拉生色譜峰均無雜質峰干擾,且陰性對照無干擾,表明方法專屬性良好。色譜圖見圖1。

1. 富馬酸2. 伏諾拉生A. 空白溶劑(流動相)B,B'. 對照品溶液C. 供試品溶液圖1 高效液相色譜圖1.Fumaric acid 2.VonoprazanA.Blank solvent(mobile phase) B,B'.Reference solution C.Test solutionFig.1 HPLC chromatograms
線性關系考察:分別精密量取伏諾拉生及富馬酸對照品貯備液各適量,加流動相逐級稀釋為質量濃度分別為100,160,200,240,300 μg / mL 的伏諾拉生系列標準溶液及34,54.4,68,81.6,102 μg / mL 的富馬酸系列標準溶液,按2.1 項下色譜條件依次進樣測定,分別以伏諾拉生及富馬酸質量濃度(X,μg / mL)對峰面積(Y)進行線性回歸,得回歸方程分別為Y伏=208 471.889 0X伏- 9 239.014 0(r= 1.000 0,n= 5)和Y富=22 211X富+9 228.5(r=0.999 9,n=5)。結果表明,伏諾拉生和富馬酸的質量濃度分別在100~300μg/mL和34~102μg/mL范圍內與峰面積線性關系良好。
檢測限與定量限確定:取線性關系考察項下質量濃度分別為100μg/mL的伏諾拉生標準溶液和34μg/mL的富馬酸標準溶液,加流動相稀釋,按2.1 項下色譜條件進樣測定,分別以信噪比(S/N)為3時的含量為檢測限、S/N為10時的含量為定量限。結果伏諾拉生檢測限為0.4 ng,定量限為1.0 ng;富馬酸檢測限為0.5 ng,定量限為1.4 ng。
精密度試驗:取2.2項下伏諾拉生及富馬酸對照品溶液各適量,按2.1 項下色譜條件連續進樣測定6 次,記錄色譜圖。結果伏諾拉生、富馬酸峰面積的RSD分別為0.48%和0.26%(n=6),表明儀器精密度良好。
穩定性試驗:取2.2項下對照品溶液和供試品溶液各適量,分別于室溫下放置0,4,8,12,24 h 時按2.1 項下色譜條件進樣測定。結果供試品溶液中伏諾拉生和富馬酸的峰面積最大變化率均為1.13%,對照品溶液中伏諾拉生和富馬酸的最大峰面積變化率分別為1.42%和1.19%,表明供試品溶液和對照品溶液在室溫下放置24 h穩定性良好。
重復性試驗:取樣品適量,精密稱定,按2.2項下方法制備供試品溶液,共6 份,按2.1 項下色譜條件依次進樣測定,按外標法以峰面積計算含量。結果伏諾拉生和富馬酸的平均含量分別為74.87%,24.65%,RSD分別為0.57%,0.55%(n=6),表明方法重復性良好。
加樣回收試驗:取樣品適量,精密稱定,按2.2項下方法制備供試品溶液,精密量取富馬酸和伏諾拉生對照品貯備液各適量,加流動相稀釋并定容,搖勻,濾過,分別作為相對限度濃度80%,100%,120%的加樣回收試驗溶液,每個濃度各平行制備3 份。按2.1 項下色譜條件進樣測定,記錄色譜圖,按外標法以峰面積計算伏諾拉生及富馬酸的含量,將理論值與測得值進行比較,并計算加樣回收率。結果見表1。
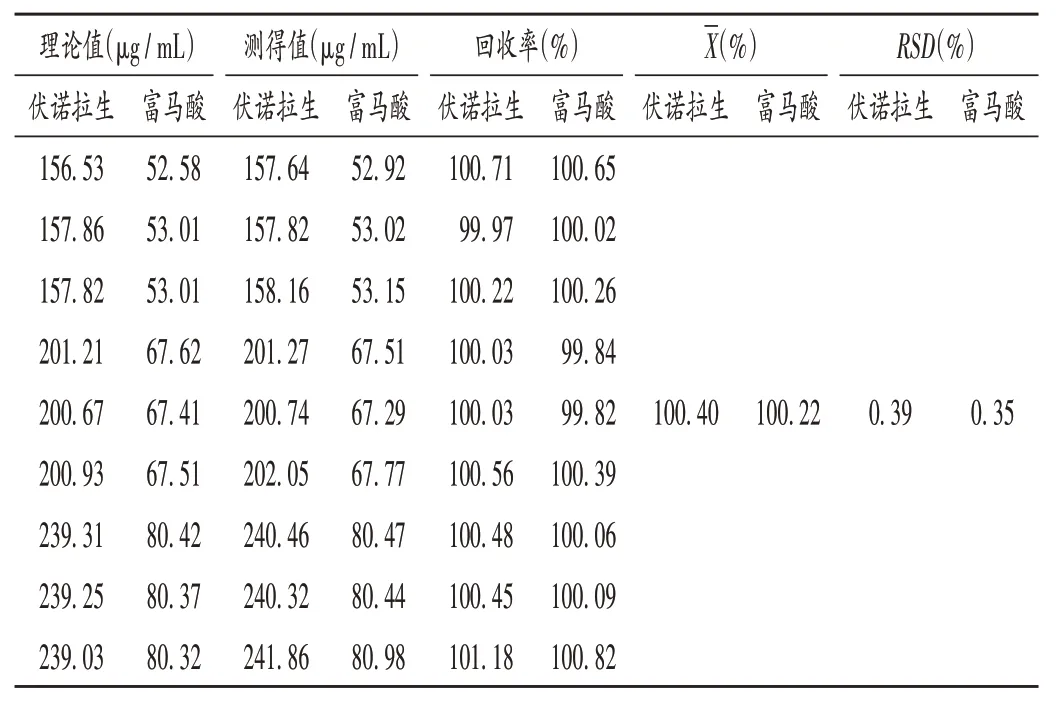
表1 加樣回收試驗結果(n=9)Tab.1 Results of the recovery test(n=9)
耐用性試驗:考察了流速(0.8,1.2 mL/min),柱溫(25,35 ℃),檢測波長(225,235 nm),流動相比例[磷酸鹽緩沖液-乙腈-甲醇(17∶6∶7,17∶7∶6,V/V/V)],磷酸鹽濃度(0.04,0.06 mol/L),流動相pH(磷酸鹽pH為6.6,7.0),色譜柱[Wondasil C18柱、Agilent 5 TC - C18(2)柱]對樣品中伏諾拉生和富馬酸含量的影響。結果顯示,色譜條件微調后,伏諾拉生和富馬酸含量與標準條件對比最大變化值分別為0.78%和0.33%,表明方法耐用性良好。
2.4 樣品中富馬酸和伏諾拉生含量測定
取不同研究階段富馬酸伏諾拉生,按2.2項下方法制備供試品溶液,按2.1 項下色譜條件進樣測定,按外標法以峰面積計算伏諾拉生和富馬酸的含量,以富馬酸與伏諾拉生的摩爾比計算成鹽率。成鹽率(%)=(富馬酸含量/ 116.07)/(伏諾拉生含量/ 345.39)×100%。結果見表2。
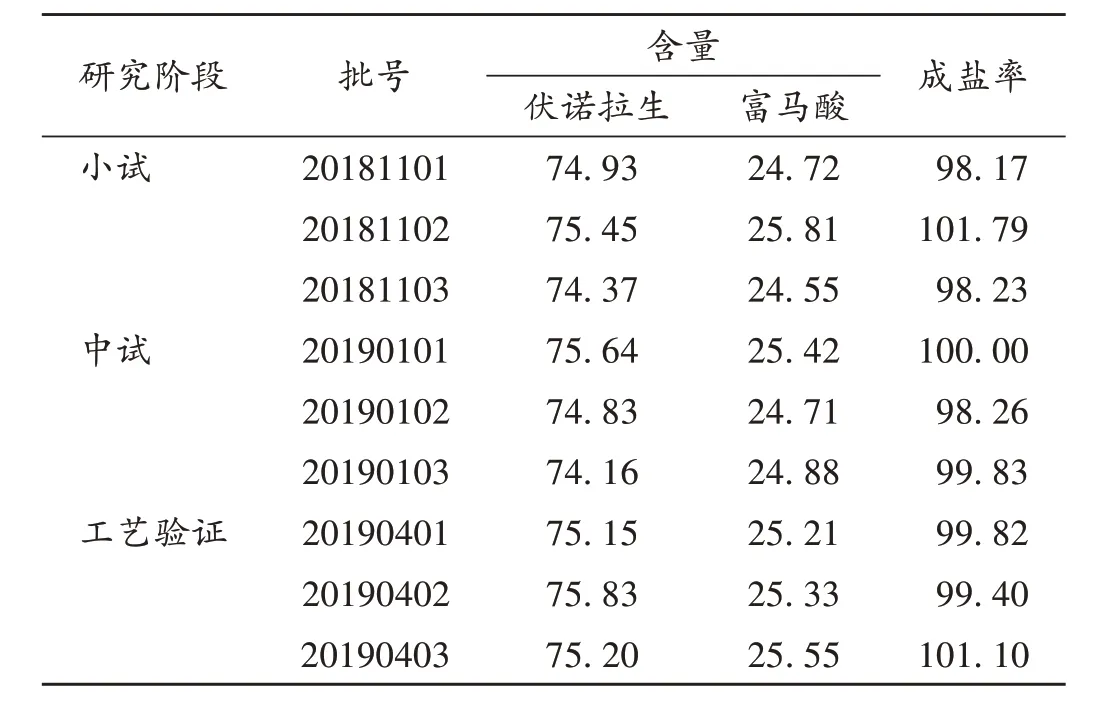
表2 樣品中富馬酸和伏諾拉生含量及成鹽率測定結果(%)Tab.2 Determination results of fumaric acid and vonoprazan content and salt - forming rate in samples(%)
2.5 富馬酸伏諾拉生差示掃描量熱曲線測定
取不同研究階段樣品各1批(批號分別為20181101<小試>、20190101<中試>、20190401<工藝驗證>),取5 mg,精密稱定,置鋁盤中,設置參數為氮氣流速20 mL/min,升溫速率10 ℃/min,測試溫度60~230 ℃。結果顯示,樣品熔融峰值范圍為207.57~208.37 ℃,焓變值(歸一化值)范圍為-210.25~-204.44 J/g,熔化時能量變化基本一致。詳見圖2。

圖2 富馬酸伏諾拉生差示掃描量熱曲線Fig.2 Differential scanning calorimetry curves of vonoprazan fumarate
3 討論
3.1 流動相選擇
伏諾拉生為弱堿性化合物,解離常數(pKa)為9.3;富馬酸為弱酸性化合物,pKa1為3.02,pKa2為4.38。對于離子型化合物,為獲得良好的峰形和保留時間的穩定,流動相pH需結合化合物pKa考慮。本研究中比較了不同pH 流動相,如pH 3.0 磷酸鹽緩沖液- 乙腈[19]、pH 6.0 磷酸鹽緩沖液-乙腈[20]、pH 6.8 磷酸鹽緩沖溶液-乙腈[23],最終選擇pH 6.8 磷酸鹽緩沖液-乙腈作為流動相。試驗中發現未知雜質干擾富馬酸檢測,經反復試驗,發現pH 6.8磷酸鹽緩沖液-乙腈-甲醇(17∶7∶6,V/V/V)為流動相時分離效果較好,雜質不干擾檢測,保留時間合適,色譜峰峰形良好。故最終選擇2.1 項下流動相。
3.2 提取溶劑及提取方法選擇
富馬酸伏諾拉生難溶于甲醇、水和乙腈[2],本研究中對不同提取溶劑和提取方法進行了考察。采用水作為提取溶劑時加樣回收率偏低,不符合要求;采用甲醇或乙腈作為提取溶劑時會產生較大空白溶劑峰,干擾富馬酸檢測,故最終選擇流動相作為提取溶劑。考察了超聲和振搖2種提取方法,并對比了不同超聲時間(10,20,30 min)。結果顯示,超聲與振搖結果無顯著差異,延長超聲時間,測定結果基本無變化,故最終選擇超聲提取20 min。
3.3 避光與不避光對比
富馬酸伏諾拉生在光照降解條件下會產生多個雜質[19]。本研究中在樣品制備及進樣過程中使用棕色瓶和白色瓶進行了對比,以確定是否需要避光操作。結果顯示,不避光條件下,樣品穩定性較差,故前處理過程及進樣過程中均需使用棕色瓶進行避光操作。
3.4 成鹽率評估
經小試、中試及工藝驗證,多批次樣品成鹽率在98.17%~101.79%范圍內,無顯著差異,說明樣品成鹽工藝穩定。富馬酸含量測定結果與理論值接近,說明成鹽完全。同時,差示掃描量熱曲線測定中,不同研究階段樣品焓變值基本一致,說明樣品成鹽工藝穩定。
3.5 方法評價
本研究中建立的方法專屬性強、準確度高、穩定性好,可為富馬酸伏諾拉生的質量控制提供參考。
