降鈣素原對脂多糖誘導的人臍靜脈內皮細胞NLRP3和caspase-1表達的影響
2023-05-27 07:22:32蔣文石丁華何艷娟陳淳媛
中國當代兒科雜志 2023年5期
蔣文 石丁華 何艷娟 陳淳媛
(1.中南大學湘雅三醫(yī)院兒科,湖南長沙 410013;2.長沙市第一醫(yī)院兒科,湖南長沙 410005)
膿毒癥是指由于宿主對感染的反應失調,而產生的危及生命的器官功能障礙[1]。目前膿毒癥是全球性的醫(yī)療保健問題,也是重癥監(jiān)護室內常見的疾病,以及感染導致死亡的主要原因,在兒童重癥監(jiān)護室內嚴重膿毒癥的全球患病率為8.2%,膿毒癥休克的病死率高達40%,甚至更高[2-3]。膿毒癥是一個炎性反應過程,血管內皮細胞損傷是膿毒癥發(fā)生發(fā)展的重要因素。在膿毒癥事件中,血管內皮細胞是炎性因子首先攻擊的目標[4-5],受病原體及內毒素等細菌產物攻擊,內皮細胞的損傷是參與膿毒癥休克、器官功能障礙及凝血功能障礙的中心環(huán)節(jié)[6-8]。
焦亡是一種依賴于半胱氨酸天冬氨酸蛋白酶(cysteinyl aspartate specific proteinase,caspase)-1、caspase-4/5介導的,以細胞質膜破裂并釋放白細胞介素-1β(interleukin-1β,IL-1β)、IL-18 等促炎性細胞因子引發(fā)細胞死亡為特點的細胞程序性死亡方式,也是宿主對抗細菌、真菌、病毒或原生動物病原體侵襲的一種方式[9-10]。細胞焦亡的本質是一種炎性宿主反應,研究表明在膿毒癥過程中,細胞焦亡一方面起到抗原提呈及清除細胞內病原體的作用,另一方面過度的焦亡激活會加重膿毒癥器官功能損害[11-12]。降鈣素原(procalcitonin,PCT)被公認為是鑒別細菌感染和病毒感染的重要生物標志物,目前作為膿毒癥的潛在生物標記物被廣泛研究[13]。最新研究表明,PCT不僅僅作為膿毒癥的重要炎性標志物,也是一種重要的炎性介質[14]。本研究以人臍靜脈內皮細胞(human umbilical vein endothelial cells,HUVECs)為研究對象,旨在探討PCT對內皮細胞焦亡相關蛋白表達的影響,為尋找膿毒癥致病機制提供新的理論依據(jù)。
1 材料與方法
1.1 材料
HUVECs 系購自美國模式培養(yǎng)物集存庫(American Type Culture Collection)。人PCT 凍干粉(用蒸餾水溶解,配制成100 μg/mL的母液備用)和LPS 凍干粉(用完全培養(yǎng)基溶解,配制成1 mg/mL的母液備用)購于美國Sigma公司。DMEM高糖培養(yǎng)基和胎牛血清購于美國Hyclone 公司。胰蛋白酶-EDTA 消化液和青鏈霉素混合液購于中國凱基生物公司。RNA 提取試劑盒(D9108A)購于美國OMEGA 公司。逆轉錄試劑盒(Rever TraAce q-PCR RT Kit)和實時熒光定量PCR試劑盒(KOD SYBR qPCR Mix)購于日本TOYOBO公司。BCA蛋白質濃度測定試劑盒和SDS-PAGE凝膠制備試劑盒購于北京碧云天生物科技有限公司。核苷酸結合寡聚化結構域樣受體蛋白3 (nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)兔多克隆抗體和caspase-1 兔多克隆抗體購于中國武漢三鷹公司。β-actin 兔多克隆抗體和羊抗兔IgG二抗購于中國博士德生物公司。
1.2 細胞培養(yǎng)
HUVECs 用含10%胎牛血清及雙抗的高糖DMEM 培養(yǎng)基于37℃、5%CO2飽和濕度培養(yǎng)箱培養(yǎng),間隔2 d 左右換液,當細胞生長達80%~90%時,用胰蛋白酶消化傳代。
1.3 實驗分組
取對數(shù)生長期HUVECs進行分組和干預,培養(yǎng)基中以濃度為1 μg/mL 的LPS 對細胞干預12 h,建立膿毒癥內皮細胞損傷模型[15],再用PCT 進行干預12 h。實驗分為兩部分:(1)正常對照組、LPS組(濃度1 μg/mL)、PCT 組(濃度10 ng/mL)及LPS+PCT 組(LPS干預后再用10 ng/mL PCT 處理);(2)正常對照組、LPS 組、LPS+PCT 不同濃度組(PCT 0.1 ng/mL 組、PCT 1 ng/mL 組、PCT 10 ng/mL組、PCT 100 ng/mL組)。
1.4 實時熒光定量聚合酶鏈反應法檢測mRNA表達水平
收集各組細胞,按RNA 提取試劑盒說明書提取總RNA,分光光度計檢測RNA 的濃度和純度。采用逆轉錄試劑盒按說明書將RNA 逆轉錄成cDNA。根據(jù)試劑盒(KOD SYBR qPCR Mix)說明書,取1 μL cDNA進行PCR擴增。PCR擴增反應條件:98℃預變性2 min;98℃變性10 s,60℃退火10 s,68℃延伸30 s,共40 個循環(huán)。以GAPDH作為內參基因,采用2-△△CT法計算目的基因mRNA 的相對表達量。引物由湖南擎科生物科技有限公司合成,引物序列見表1。實驗獨立重復3次。
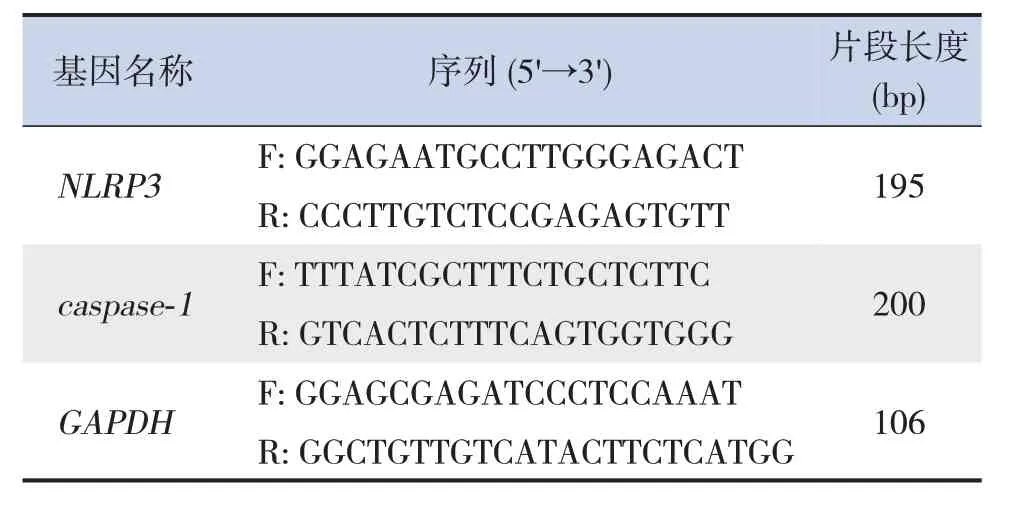
表1 NLRP3、caspase-1和GAPDH基因引物序列
1.5 Western blot法檢測蛋白表達水平
收集各組細胞,提取細胞總蛋白,BCA蛋白定量試劑盒測定蛋白濃度。蛋白煮沸變性,根據(jù)SDS制膠試劑盒說明書制膠,蛋白凝膠電泳后將蛋白轉移至PVDF膜。用5%的脫脂牛奶封閉2 h,加入1∶1 000稀釋的兔抗人NLRP3、兔抗人caspase-1、兔抗人β-actin一抗4℃振蕩孵育過夜。PBST漂洗3次,加入1∶5 000 稀釋的羊抗兔二抗室溫振蕩孵育2 h,PBST 漂洗3 次,ECL 化學發(fā)光顯色。使用Image J 8.0 軟件對蛋白條帶進行灰度值分析。實驗獨立重復3次。
1.6 統(tǒng)計學分析
采用SPSS 21.0 統(tǒng)計軟件對數(shù)據(jù)進行統(tǒng)計學分析,符合正態(tài)分布的計量資料采用均數(shù)±標準差(±s)表示。多組間比較采用單因素方差分析,組間兩兩比較采用LSD-t檢驗。P<0.05為差異有統(tǒng)計學意義。
2 結果
2.1 PCT干預對LPS誘導的HUVECs中NLRP3、caspase-1 mRNA及蛋白表達的影響
與正常對照組比較,LPS 組、PCT 組及LPS+PCT組NLRP3、caspase-1 mRNA及蛋白表達均上調(P<0.05);與LPS 組比較,LPS+PCT 組NLRP3、caspase-1 mRNA 及蛋白表達下調(P<0.05);LPS+PCT組與PCT組各指標的比較差異均無統(tǒng)計學意義(P>0.05)。見表2、圖1。
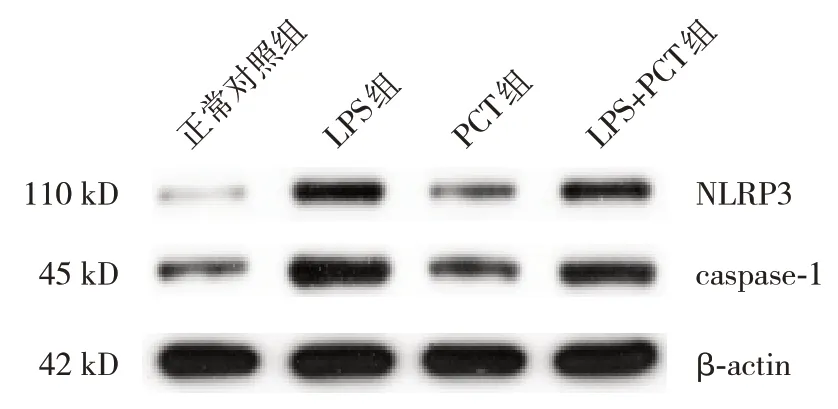
圖1 Western blot法檢測各組NLRP3、caspase-1蛋白表達電泳圖
表2 各組細胞NLRP3、caspase-1 mRNA及蛋白表達水平 (± s,n=3)
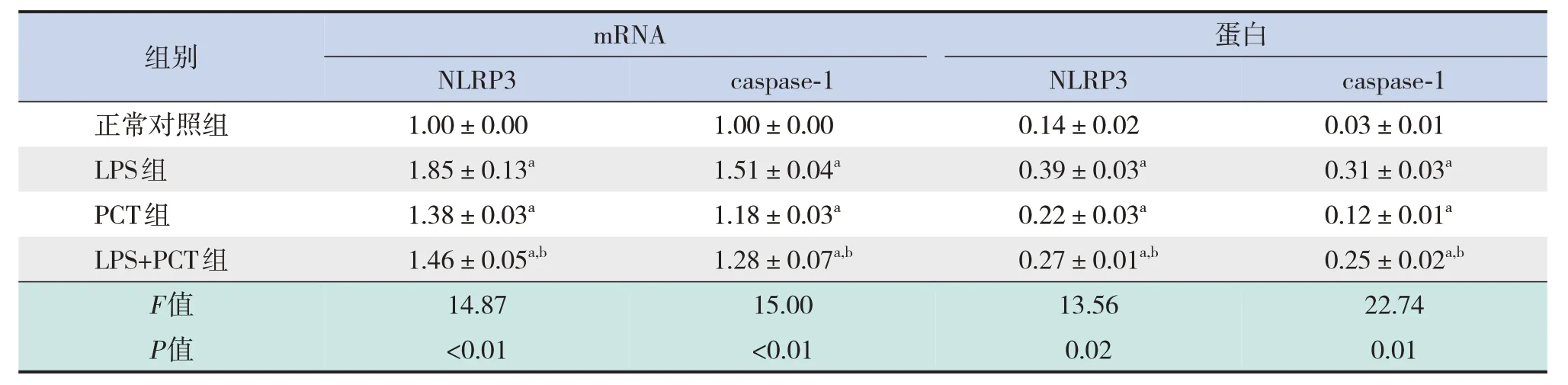
表2 各組細胞NLRP3、caspase-1 mRNA及蛋白表達水平 (± s,n=3)
注:[LPS]脂多糖;[PCT]降鈣素原;[NLRP3]核苷酸結合寡聚化結構域樣受體蛋白3;[caspase-1]半胱氨酸天冬氨酸蛋白酶-1。a示與正常對照組比較,P<0.05;b示與LPS組比較,P<0.05。
mRNA組別正常對照組LPS組PCT組LPS+PCT組F值P值蛋白NLRP3 1.00±0.00 1.85±0.13a 1.38±0.03a 1.46±0.05a,b 14.87<0.01 caspase-1 0.03±0.01 0.31±0.03a 0.12±0.01a 0.25±0.02a,b 22.74 0.01 caspase-1 1.00±0.00 1.51±0.04a 1.18±0.03a 1.28±0.07a,b 15.00<0.01 NLRP3 0.14±0.02 0.39±0.03a 0.22±0.03a 0.27±0.01a,b 13.56 0.02
2.2 不同濃度PCT 干預對LPS 誘導的HUVECs中NLRP3、caspase-1 mRNA及蛋白表達的影響
與正常對照組比較,LPS組及PCT不同濃度組NLRP3、 caspase-1 mRNA 及蛋白表達均上調(P<0.05);與LPS 組 比 較,PCT 不 同 濃 度 組NLRP3、 caspase-1 mRNA 及蛋白表達均下調(P<0.05),隨著PCT 濃度增加,NLRP3、caspase-1 mRNA及蛋白表達逐漸下調,不同濃度的PCT各組比較差異有統(tǒng)計學意義(P<0.05)。見表3、圖2。

圖2 Western blot法檢測各組NLRP3、caspase-1蛋白表達電泳圖
表3 各組細胞NLRP3、caspase-1 mRNA及蛋白表達水平 (± s,n=3)
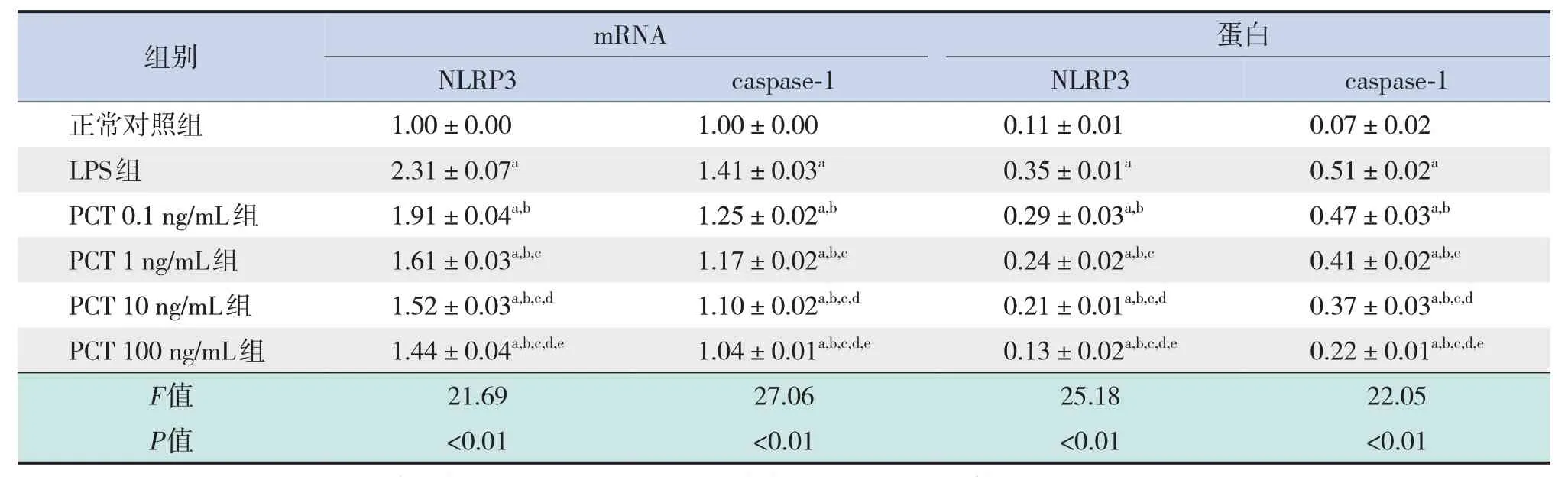
表3 各組細胞NLRP3、caspase-1 mRNA及蛋白表達水平 (± s,n=3)
注:[LPS]脂多糖;[PCT]降鈣素原;[NLRP3]核苷酸結合寡聚化結構域樣受體蛋白3;[caspase-1]半胱氨酸天冬氨酸蛋白酶-1。a 示與正常對照組比較,P<0.05;b 示與LPS 組比較,P<0.05;c 示與PCT 0.1 ng/mL 組比較,P<0.05;d 示與PCT 1 ng/mL 組比較,P<0.05;e示與PCT 10 ng/mL組比較,P<0.05。
mRNA組別正常對照組LPS組PCT 0.1 ng/mL組PCT 1 ng/mL組PCT 10 ng/mL組PCT 100 ng/mL組F值P值蛋白caspase-1 0.07±0.02 0.51±0.02a 0.47±0.03a,b 0.41±0.02a,b,c 0.37±0.03a,b,c,d 0.22±0.01a,b,c,d,e 22.05<0.01 caspase-1 1.00±0.00 1.41±0.03a 1.25±0.02a,b 1.17±0.02a,b,c 1.10±0.02a,b,c,d 1.04±0.01a,b,c,d,e 27.06<0.01 NLRP3 1.00±0.00 2.31±0.07a 1.91±0.04a,b 1.61±0.03a,b,c 1.52±0.03a,b,c,d 1.44±0.04a,b,c,d,e 21.69<0.01 NLRP3 0.11±0.01 0.35±0.01a 0.29±0.03a,b 0.24±0.02a,b,c 0.21±0.01a,b,c,d 0.13±0.02a,b,c,d,e 25.18<0.01
3 討論
在膿毒癥事件中,血管內皮細胞是該過程的主要靶細胞,內皮細胞與炎性介質、先天性免疫成分及凝血系統(tǒng)處于動態(tài)平衡狀態(tài)來協(xié)調宿主的反應。血管內皮細胞的損傷、功能障礙在膿毒癥的一系列不良后果中發(fā)揮著重要作用[16-17]。焦亡是依賴于caspase 激活的另一種細胞程序性死亡的方式,是依賴NLRP3 炎性小體、caspase-1 及Gasdermin D (GSDMD) 蛋白活化后促進IL-1β、IL-18等促炎性物質成熟及釋放的促炎過程[10,18-19]。細胞焦亡破壞體內受感染的細胞,促使感染細胞內病原體釋放,同時釋放一系列炎性因子,從而被免疫細胞識別、吞噬及殺滅,在機體內起到抗原提呈及清除細胞內病原體的作用[20-21]。因此,適度的細胞焦亡是保護機體抵御病原微生物感染的重要方式,但是過度的細胞焦亡激活可能會加重膿毒性休克和膿毒癥器官損害。血管內皮細胞的炎性損傷是膿毒癥病理過程中的關鍵,而細胞焦亡是感染性疾病中的重要調節(jié)機制。在膿毒癥急性肺損傷機制的研究中表明,GSDMD 蛋白由LPS刺激的單核細胞釋放,與NLRP3炎性小體活化后的caspase-1 結合形成微粒,GSDMD 蛋白被caspase-1 切割后活化,活化后的GSDMD 蛋白在肺血管內皮細胞上打孔,破壞細胞膜的完整性,對內皮細胞產生損傷[22]。這提示血管內皮細胞的焦亡可能是膿毒癥疾病過程重要病理機制之一。PCT的血清濃度被認為是全身感染性疾病的嚴重程度及預后的生物學標志物。而目前研究發(fā)現(xiàn),PCT本身存在細胞毒性,且有研究表明在膿毒癥肝衰竭的過程中其可能擔任了炎性介質的重要角色[23-24]。細胞焦亡的本質是促炎過程,而膿毒癥的發(fā)生發(fā)展也是一個炎癥放大的過程,PCT作為膿毒癥過程中的重要生物學標志物,在膿毒癥細胞焦亡機制中是促炎因子或保護因子,目前尚不清楚。
本研究發(fā)現(xiàn),在LPS誘導的膿毒癥內皮細胞炎癥損傷模型中,與正常對照組比較,LPS組焦亡相關蛋白NLRP3 及caspase-1 mRNA、蛋白表達均上調,說明LPS誘導的細胞焦亡參與了膿毒癥發(fā)病機制。有研究表明,在盲腸結扎穿刺誘導的膿毒癥心肌功能障礙的小鼠模型中,小鼠心肌細胞勻漿中的NLRP3 及caspase-1 mRNA 表達水平增高,同樣在體外試驗中證實,將小鼠心肌細胞暴露在LPS中,NLRP3 及caspase-1 mRNA 表達水平也增高,本研究結果與之一致。而隨著NLRP3 炎性小體的激活,IL-1β 及IL-18 等炎癥因子也隨即增高[25]。以上結果提示,膿毒癥時細菌或細菌產物的刺激激活了NLRP3 炎性小體,從而進一步介導了炎性反應。在心肌缺血-再灌注損傷引起的無菌性炎癥的研究中發(fā)現(xiàn),在心肌梗死早期NLRP3 炎性小體激活,進一步激活caspase-1,募集循環(huán)中的白細胞對損傷細胞進行吞噬,最后對心肌細胞的缺血損傷起到了一定的修復作用[26]。因此,細胞焦亡在某些炎癥過程中起到了一定的保護作用,但過度的炎性物質釋放可加重組織損傷。
本研究用不同濃度的PCT (0.1、1、10、100 ng/mL)對LPS 誘導的膿毒癥HUVECs 模型干預12 h,發(fā) 現(xiàn)PCT 使 正 常HUVECs 中NLRP3、caspase-1 表達上調,而對LPS 誘導的HUVECs 中NLRP3、caspase-1表達有抑制作用,提示PCT對正常細胞焦亡和膿毒癥模型細胞焦亡有不同作用機制。Sauer 等[23]在膿毒癥肝衰竭的體外實驗中發(fā)現(xiàn),將人肝癌細胞暴露在濃度為0.01~50 ng/mL 的PCT中會誘導細胞壞死,并降低代謝活性、細胞完整性、合成和解毒能力,而同時使用L929 成纖維細胞獲得同樣的實驗結果。在危重患者研究中發(fā)現(xiàn),PCT的升高促使了線粒體功能的障礙,使細胞代謝發(fā)生改變,同時促進了內皮細胞上促炎介質的釋放,激活單核細胞、巨噬細胞和T細胞等免疫細胞,對內皮細胞屏障功能產生損害[27]。以上結果提示,PCT通過啟動細胞焦亡、壞死或直接對細胞產生損傷,存在一定細胞毒性,在炎癥介導的過程中起促進作用。PCT對LPS干預后HUVECs中NLRP3 及caspase-1 表達又有一定抑制作用,并呈濃度依賴性,提示PCT 在膿毒癥細胞模型中對細胞焦亡又有抑制作用。Hoffmann 等[28]發(fā)現(xiàn)膿毒癥休克的血管平滑肌細胞模型中,PCT在轉錄水平上抑制LPS 介導的腫瘤壞死因子-α(tumor necrosis factor-α,TNF-α)合成。將人全血細胞與LPS 和PCT同時孵育可抑制LPS誘導的TNF-α產生[29]。以上結果表明PCT 在一定程度上通過抑制焦亡或通過減少TNF-α產生抑制炎癥過程,PCT作為一種炎癥調節(jié)劑,在LPS、TNF-α 和干擾素γ 等激動劑引發(fā)的炎癥反應過程中起重要作用[30]。因此,PCT可使正常HUVECs 中NLRP3、caspase-1 表達上調,而使LPS誘導的HUVECs中NLRP3、caspase-1表達下調,提示PCT 可能通過細胞焦亡參與膿毒癥發(fā)病機制,對正常和病理HUVECs 的NLRP3、caspase-1 表達調控機制可能存在差異,在膿毒癥中PCT 可能具有致炎和抗炎雙重作用,其作用機制及介導的通路,需進一步探討。
