廣西合浦珠母貝3個不同群體遺傳關系分析
2023-05-18 05:00:08彭慧婧鄭德斌張守都
水產科學 2023年3期
鄒 杰,彭慧婧,鄭德斌,張守都
( 1.廣西科學院,廣西 南寧 530000; 2.廣西海洋研究所有限責任公司,廣西 北海 536000; 3.天津渤海水產研究所,天津 300457; 4.山東省海洋科學研究院,山東 青島 266000 )
合浦珠母貝(Pinctadafucatassp.martensii),又稱馬氏珠母貝,是我國培育海水珍珠的主要貝類。廣西為合浦珠母貝主產地和南珠發源地,近年來,養殖環境日漸劣化、養殖群體生長性狀衰退、繁育個體趨小型化等因素,均限制了南珠產業的健康可持續發展。由于重新選育合浦珠母貝優良品種所需成本高、時間長,為適應新的養殖形勢和環境變化,需要快速改良廣西合浦珠母貝的種質資源,雜交育種是能夠短時間內提升種質資源的方式之一。有關不同地理群體合浦珠母貝雜交研究較多[1-2],其具有一定的雜種優勢[3],但關于廣西合浦珠母貝地理群體內雜交的研究較少。要加快廣西合浦珠母貝群體雜交育種應用,首先要了解廣西合浦珠母貝主要群體的遺傳背景。目前廣西合浦珠母貝地理群體內經人工繁育或選育分化成3個主要群體:選育群體“海選1號”、養殖群體和野生繁育群體。選育群體是以廣西潿洲島海域野生群體選育出的優質新品種,已在廣東和廣西應用多年,生長性狀具有顯著優勢[4];養殖群體則是合浦傳統南珠養殖區繼代繁育養殖群體,存在一定的抗逆優勢;野生群體表型分化較大,遺傳資源可能具有潛在的復雜多態性。這3個分化群體可作為廣西合浦珠母貝群體雜交親本的潛在來源群。由于長期混雜養殖,3個分化群體間理論上存在基因交流,在進行雜交育種時應首先掌握選擇親本群體的真實遺傳分化狀況,分析不同群體的近交水平遺傳分化程度,進而提升雜交育種的預期效果。鑒于遺傳標記多態性可有效分析合浦珠母貝的遺傳差異[5-6],筆者通過對3個群體中的適齡種貝作為樣本進行簡化基因組測序,通過單核苷酸多態標記解析3個樣本群間的遺傳關系,從一定程度上反映3個群體的遺傳背景,探索出不同群體間的最佳雜交組合方式,為廣西合浦珠母貝雜交育種中的親本選擇提供參考。
1 材料與方法
1.1 材料處理
分別從野生群體(F1)、選育群體(“海選1號”F4)和養殖群體中隨機挑選15個2齡種貝,組成3個樣本群體,分別采集軟體部-80 ℃保存(3個月內),利用1端EcoRⅠ(G^AATTC)和2端NlaⅢ(Hin1Ⅱ,CATG^)進行雙酶切,將質檢合格的DNA樣品500 ng采用ddRAD建庫方式構建長度在300~500 bp的雙端測序文庫,基于Illunima HiSeq 2500高通量測序。
1.2 數據處理
1.2.1 數據質控
對片段測序得到的原始讀長進行數據評估,得到各個樣品的原始數據,通過堿基質量值(Q)進行質控獲得高質量數據。
堿基質量值(Q)與錯誤率(Re)的關系式為:
Q=-10lgRe
1.2.2 信息分析
將序列讀長使用BWA比對到近緣[7]參考物種覆瓦珠母貝(P.imbricata)染色體上(GCA_002216045.1_PinMar1.0),根據高質量數據在參考染色體的定位結果,未掛載上染色體的拼接序列共同作為參考基因組,使用Picard(0.7.17-r1188)的Mark Duplicate工具去除重復,屏蔽PCR重復數據的影響,使用GATK進行變異檢測,變異位點質量值重新校正,后進行測序深度過濾,平均測序深度為5×,最小等位基因頻率為0.05,樣品信息完整度為0.70,變異位點質量值Q>30,過濾獲取可靠變異結果形成單核苷酸多態標記,用于后續分析。基于單核苷酸多態標記,對序列雙端比對基因組率超90%個體,通過軟件GCTA(1.92.1,http://cnsgenomics.com/software/gcta/#Overview)進行主成分分析,得到樣品的主成分,運用R軟件繪制主成分分析圖,利用軟件FastTree(2.1.9)中的極大似然法構建極大似然進化樹;使用軟件PopLDdecay(https://github.com/BGI-shenzhen/PopLDdecay)進行連鎖不平衡衰減分析,參數為:-OutPairLD5;在軟件vcftools(0.1.16)中,參數設為以100 kb為一個滑動窗口,10 kb為步長取一個區域,計算群體在這個區域內的群體分化指數(Fst);在R軟件(R包:detectRUNS)中,設定每一個滑動窗口至少要有20個單核苷酸多態標記,滑動窗口閾值使用默認參數0.05,每一個滑動窗口中允許的相反基因型數目為1個,每一個滑動窗口中允許丟失的基因型為1個,組成長純合片段(ROH)之間的單核苷酸多態標記的最大間隔為1 Mb,長純合片段的最小長度設為0.25 Mb,基于參考染色體統計長純合片段,基于每條染色體分析近交系數(FROH)。
FROH=∑LROH/Lgenome
式中,∑LROH為染色體上長純合片段的長度之和,Lgenome為染色體的物理總長度。
2 結果與分析
2.1 測序與參考染色體比對結果
45個樣品共產生49.79 Gb高質量數據,對測序數據進行質控評估結果見表1,質控平均Q30為92.17%,3群體GC含量34.59%~35.04%,低于AT含量。組裝參考基因組大小為990 984 031 bp,未掛載參考染色體的基因組大小為137 058 386 bp,樣品平均比對讀長為768 337 bp,平均覆蓋基因組大小為85 103 126 bp,平均覆蓋深度為13.84,平均基因組覆蓋深度為0.99,選育群體的平均比對、雙端比對讀長均較低。
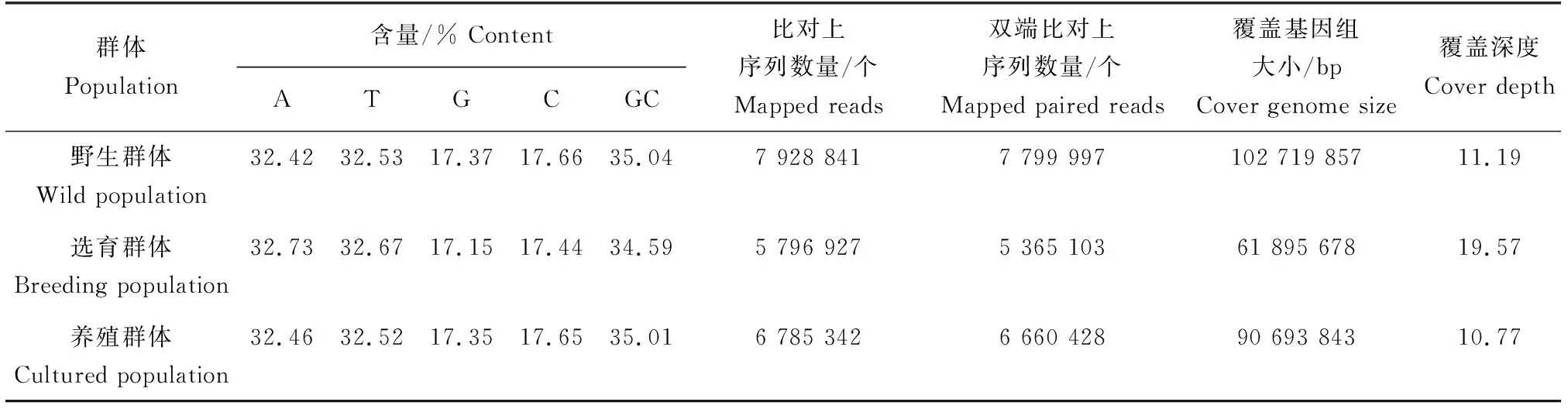
表1 不同群體測序堿基分布和與參考基因組比對結果(均值)Tab.1 Distribution and comparison of sequencing bases with reference genome in different populations (mean)
2.2 單核苷酸多態與群體信息統計
樣品與參考基因組之間單核苷酸多態標記檢測平均轉換數量為206 641個,平均顛換數量為184 471個(圖1a),轉換/顛換為1.12,轉換發生率高于顛換,平均雜合類型單核苷酸多態標記數量為151 042個,野生群體雜合程度最高,平均純合單核苷酸多態標記數量為240 070個,選育群體平均純合單核苷酸多態標記數量高于野生群體和養殖群體。群體單核苷酸多態標記過濾后,1、3、9和10號染色體單核苷酸多態標記數量較多(圖1b),以20 kb為一個窗口,統計窗口內的單核苷酸多態標記數量,單核苷酸多態標記在染色體上的密度分布較均勻(圖1c),染色體單核苷酸多態標記較多與其長度相關。統計單群體內的雙等位基因型群體遺傳指標平均值,群體信息統計見表2,群體內單核苷酸多態標記統計數量超過58萬個,選育群體的觀測雜合度最低,平均近交系數(Fis)與養殖群體相近,野生群體觀測雜合度最高,近交系數(Fis)最低,野生群體相對具有較好的群體遺傳多態。
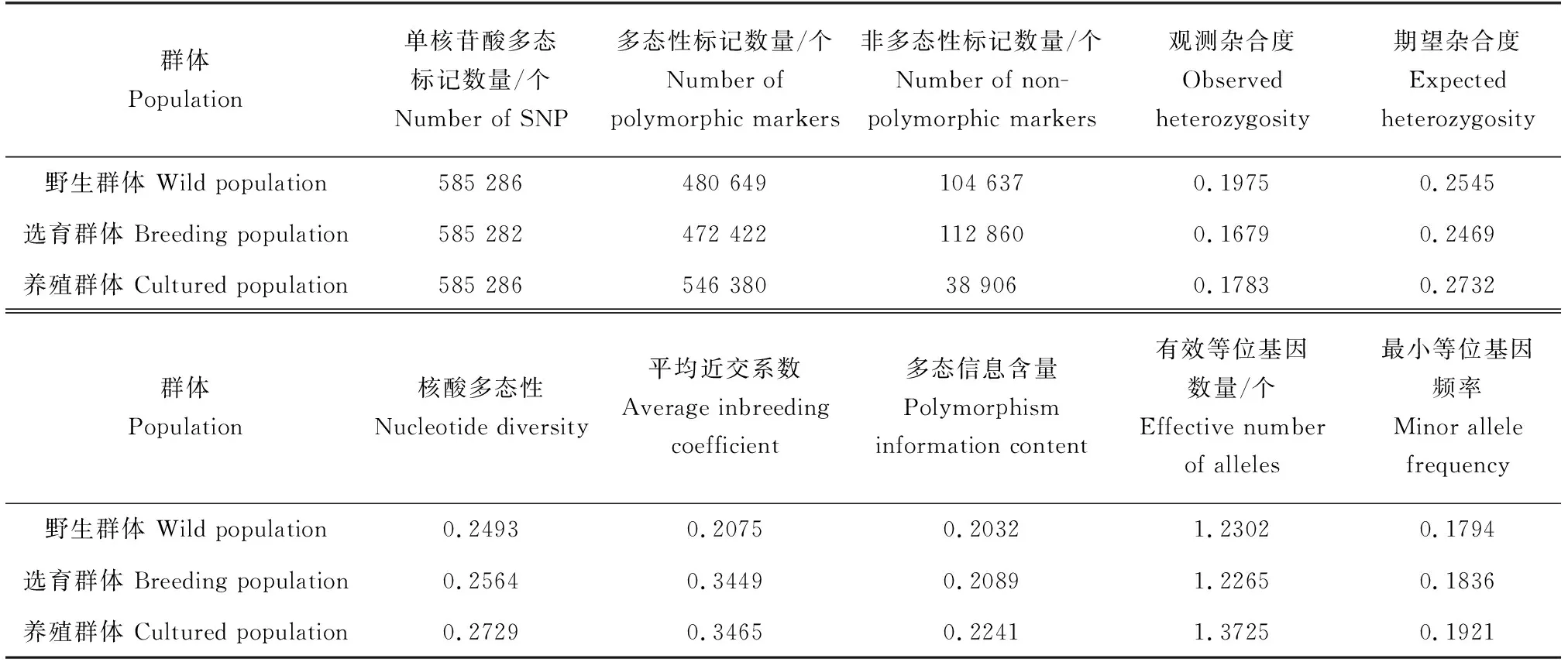
表2 群體遺傳信息統計Tab.2 The statistics of population genetic information
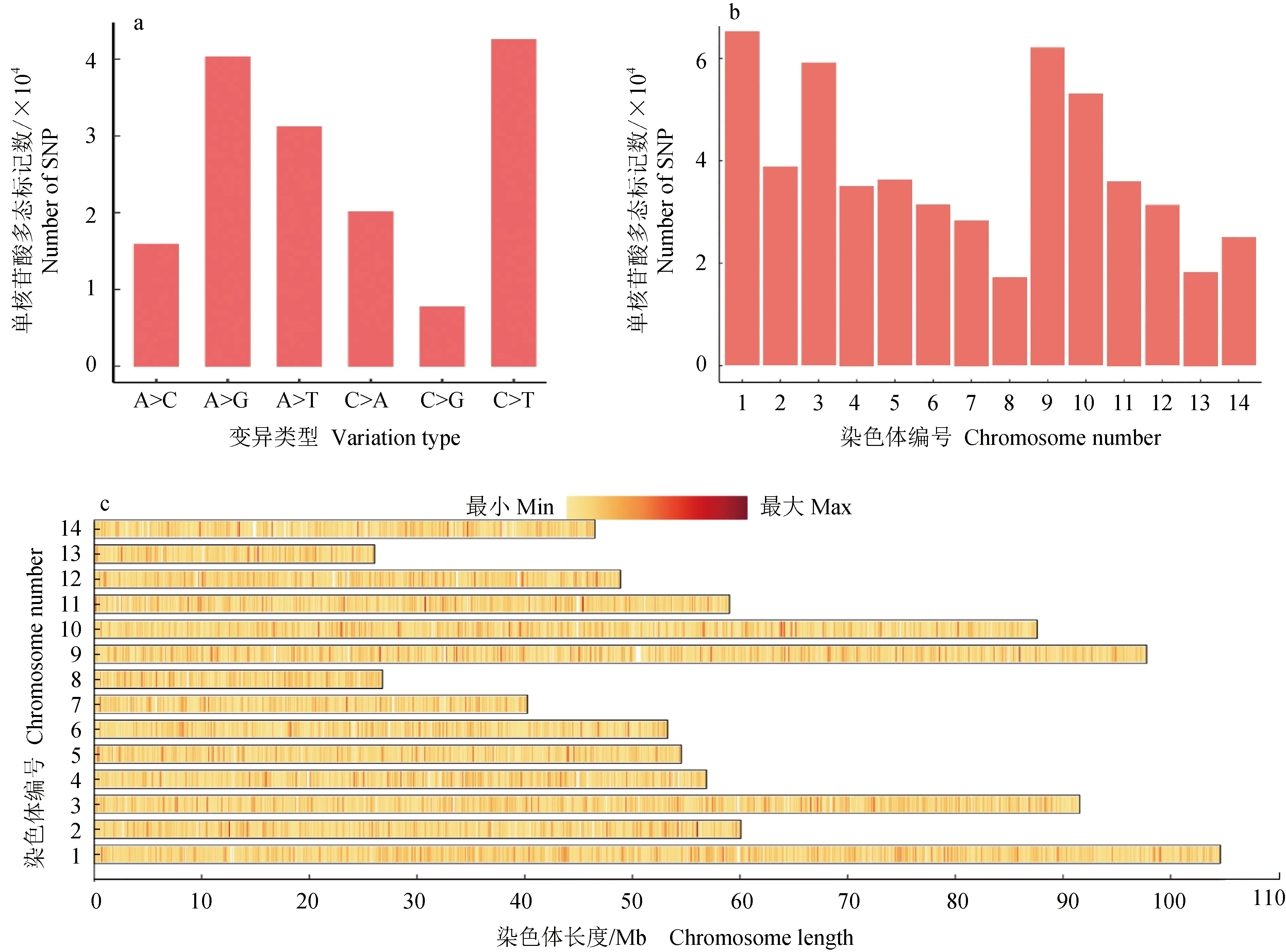
圖1 單核苷酸多態標記統計Fig.1 SNP statisticsa.單核苷酸多態類型變異數量統計;b.染色體單核苷酸多態標記數量統計;c.染色體單核苷酸多態標記密度分布.a.SNP type variation statistics; b.statistics of chromosome SNPs; c.distribution of chromosome SNP density.
2.3 群體分化分析
利用與參考染色體高比對(>90%)樣品的單核苷酸多態標記進行系統進化樹(圖2)和主成分(圖3)分析,結果顯示,選育群體與養殖群體部分樣品親緣關系較近,預示選育群體和養殖群體可能有部分相同的親本來源,主成分分析基本將樣品分成3個類群,主成分1顯示野生群體分類較好,主成分2顯示選育群體和養殖群體存在群體交雜,分析結果與進化樹分析一致。通過計算區域平均遺傳分化指數(Fst),野生群體-選育群體、野生群體-養殖群體和選育群體-養殖群體的遺傳分化指數均值分別為0.068、0.065和0.030,選育群體和養殖群體之間的遺傳分化指數最低,多態性相近,進一步表明部分親本來源相同的可能性較大。
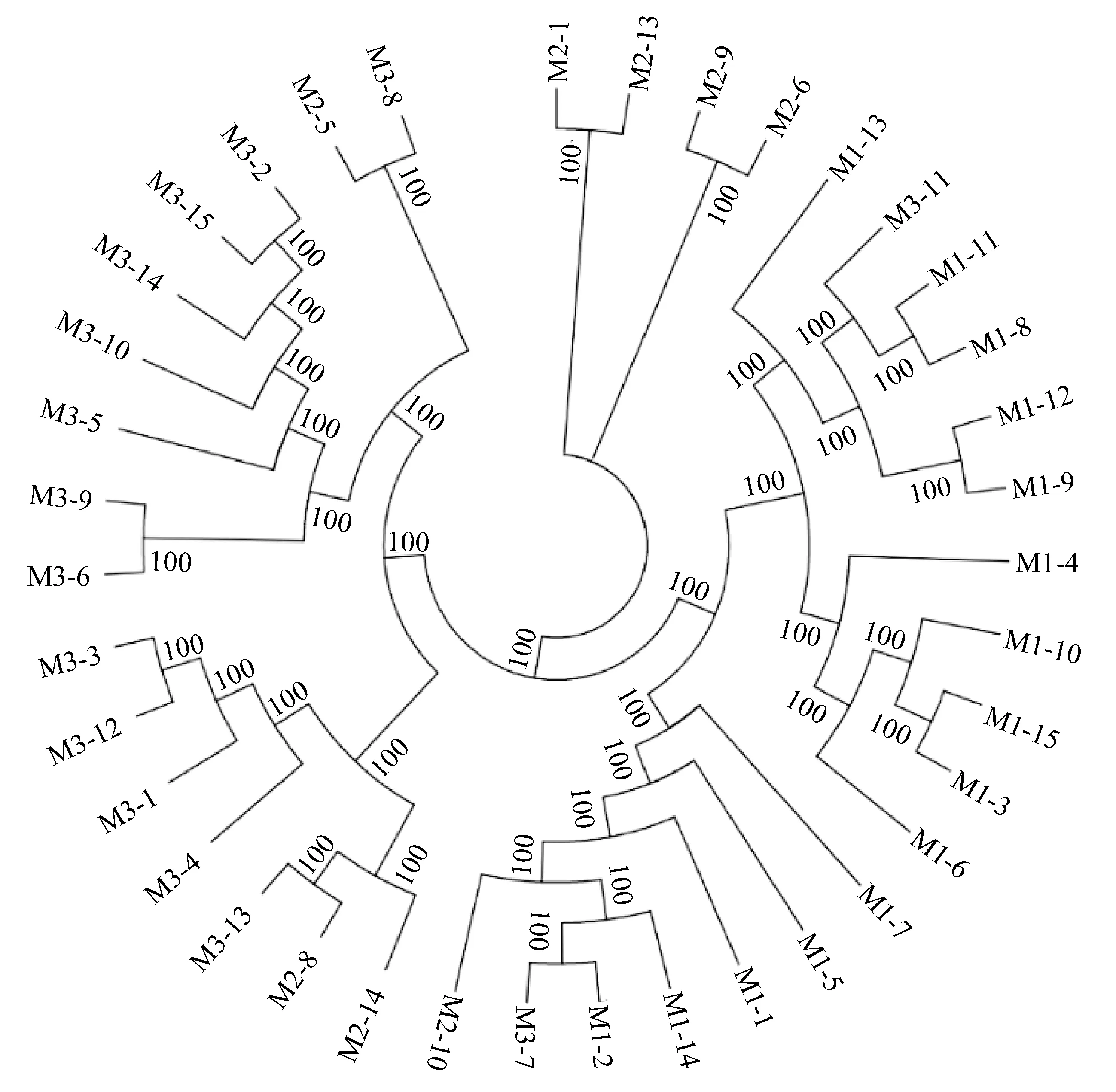
圖2 系統進化樹Fig.2 Phylogenetic tree個體編號中,M1表示野生群體,M2表示選育群體,M3表示養殖群體.In the individual number, M1 represents wild population, M2 represents breeding population and M3 represents cultured population.
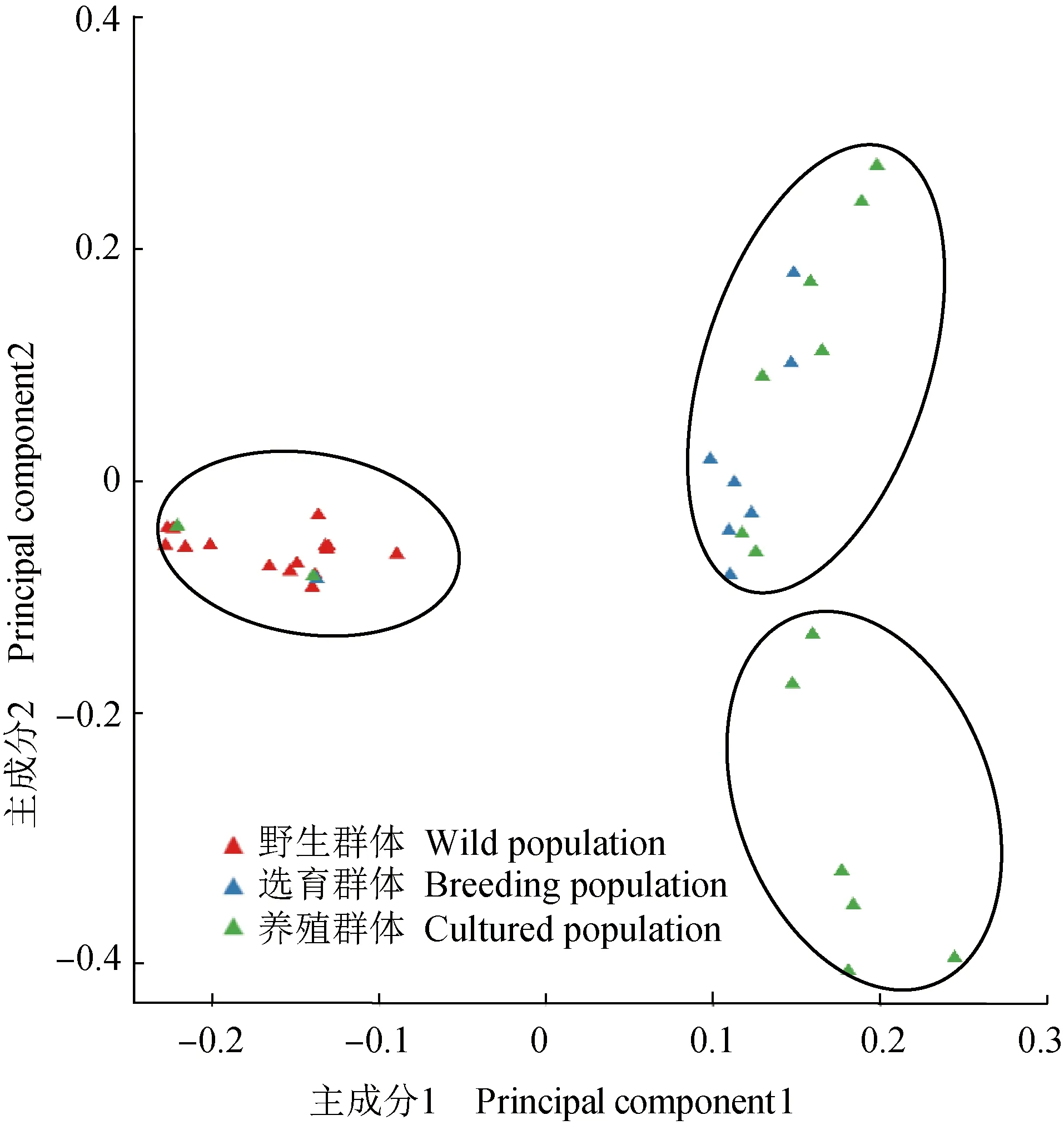
圖3 主成分分析Fig.3 Analysis of principal component analysis
2.4 長純合片段與連鎖不平衡衰減分析
野生群體、選育群體和養殖群體的平均長純合片段數分別為59.1、49.5個和56.6個,長純合片段平均長度分別為0.312、0.313Mb和0.311Mb,平均單核苷酸多態標記含量為59、50個和73個,1號和10號染色體長純合片段平均含量較多,為6.5個,7號最少,為1.9個。基于長純合片段分布和比例計算的近交系數(FROH)見圖4,野生群體、選育群體和養殖群體的近交系數(FROH)分別為0.0149~0.0240、0.0159~0.0201和0.0170~0.0246,14號染色體近交系數(FROH)最高,野生群體近交系數(FROH)略高于選育群體和養殖群體,3個群體近交系數(FROH)均低于0.060。整個染色體內的連鎖不平衡衰減結果見圖5。一般把連鎖不平衡系數(r2)降低至0.1~0.2時的遺傳距離認為是該物種的衰減距離。r2為0.2時,野生群體、選育群體和養殖群體的平均連鎖不平衡的距離為0.4、0.4 kb和0.2 kb;r2降為0.1時,野生群體、選育群體和養殖群體的平均連鎖不平衡距離增至40.5、>300.0 kb和4.0 kb。衰減速度為養殖群體>野生群體>選育群體,養殖群體具有較高的群體內核酸多態性(表2),結果與其衰減速度最快相一致,選育群體由于連續多年人工選育,群體內核酸多態性較低,衰減速度最慢。
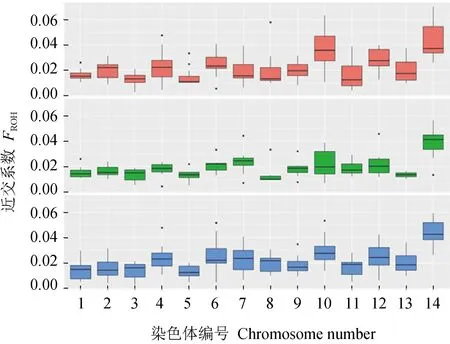
圖4 不同染色體近交系數統計Fig.4 Statistics of FROH in different chromosomes紅色代表野生群體,綠色代表選育群體,藍色代表養殖群體.Wild population is shown in red color, breeding population is shown in green color and cultured population is shown in blue color.
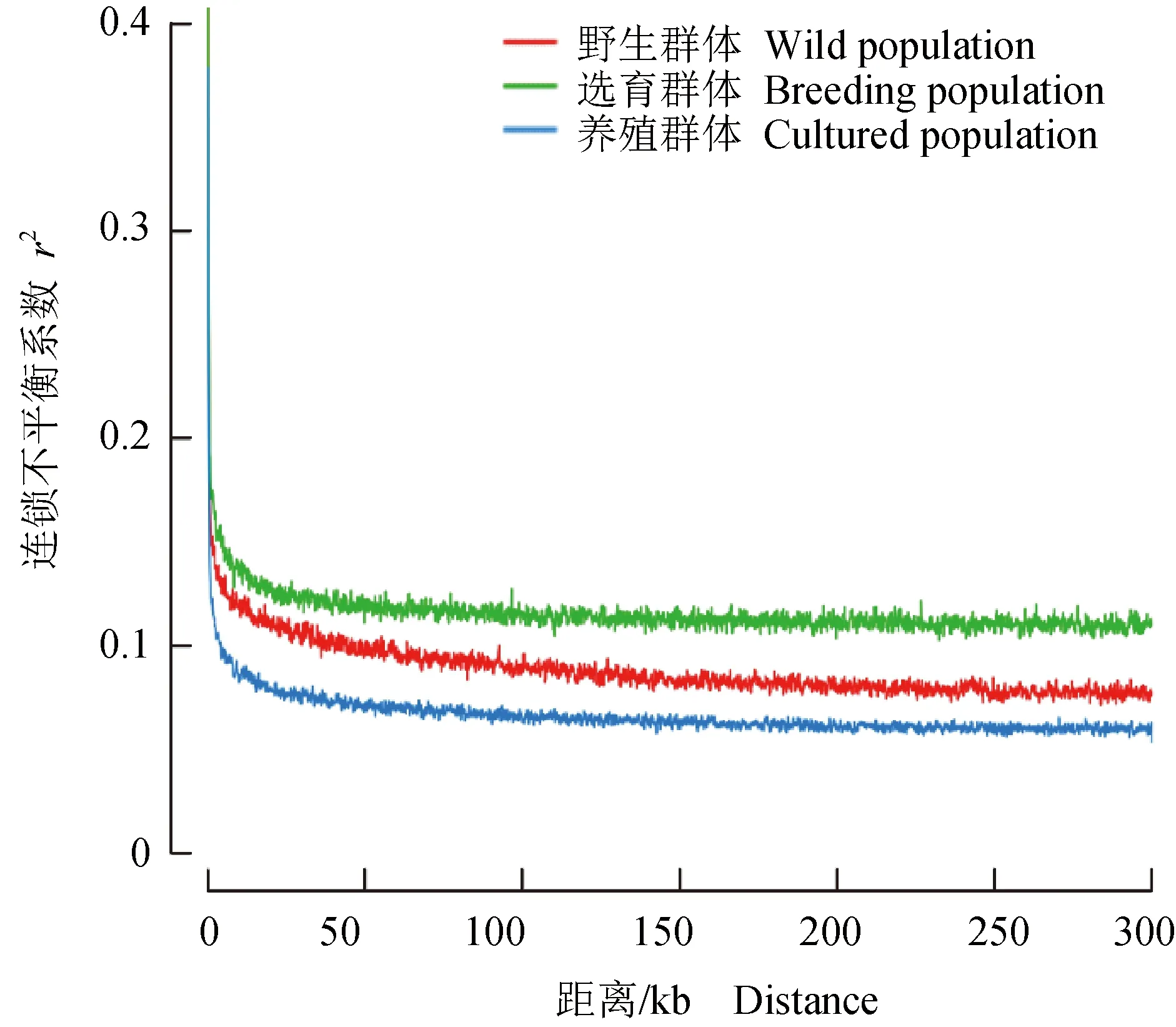
圖5 不同群體連鎖不平衡衰減Fig.5 Linkage disequilibrium decay in different populations
3 討 論
3.1 群體分化與雜交潛力分析
為獲取有效的雜種優勢,雜交親本應選用不同分化亞群或具有較好遺傳資源的群體(系)或品系,并從來源群體中選擇具備一定表型優勢的親本,野生群體作為來源時應該純化親本[8]。合浦珠母貝養殖性狀中生長速度[9]和存活率均是影響產珠的重要選擇性狀,在難以估計存活性狀的情況下,生長相關表型性狀成為親本的首選性狀,筆者均利用人工繁選群體作為親本的來源群,保證了親本的純化和優勢性狀親本的選擇,方便準確分析適合作為雜交親本的遺傳信息。
本研究中,3個群體經過群體多態指數比較、主成分分析交叉位點和系統進化樹分析得出,選育群體和養殖群體群體分化程度最低。遺傳分化指數是衡量群體間的遺傳分化程度的重要指標,當0<遺傳分化指數<0.05時,表明各群體之間分化很小,幾乎可以忽略;當遺傳分化指數>0.05時,各群體之間才存在中度以上的遺傳分化[10]。馬氏珠母貝的不同殼色家系間遺傳分化指數為0.113~0.793[11],表明各個家系之間存在極大的分化,可能與連續多年的家系選育以及近交引起家系內基因頻率發生較大變化有關;在馬氏珠母貝近交與雜交4個家系間的平均遺傳分化指數為0.1049[12],表明4個家系間存在中度的遺傳分化,同雜交和近交的不同交配方式對基因頻率改變存在重要關系;本研究的平均遺傳分化指數相對較低,其中野生群體-選育群體和野生群體-養殖群體的遺傳分化指數均值分別為0.068和0.065,表明野生群體-選育群體、野生群體-養殖群體之間存在中等程度的分化,與選育群體的連續多年選育,以及養殖群體在養殖中被有意識作為一個獨立群體繁育有關。選育群體-養殖群體的遺傳分化指數均值為0.030,表明選育群體和養殖群體間幾乎沒有遺傳分化,表明本研究中的2個群體間的基因頻率差別不大,群體來源相同或部分個體來源相同的可能性較大,這可能與本研究選用來源群體的粗放保種以及生產中的不當操作串種有較大關系。廣西合浦珠母貝產業受養殖環境的影響,導致傳統養殖群體萎縮,而隨著選育群體在廣西的推廣應用力度加大,一定程度上造成傳統養殖群體與選育群體的混雜并存,且選育群體本身具有優勢生長表型性狀[4],所以在選擇同質種貝樣本時,樣本同源的概率較大。連鎖不平衡衰減速度能夠反映群體選擇強度或群體多態性[13],野生群體受人工選擇的壓力較小,養殖群體雖然多態性標記數量(546 380個)最多,但與選育群體分化程度偏低。為了引入更多的優秀基因,在沒有專門化群系條件下,進行快速育種時可優先考慮野生群體×選育群體或野生群體×養殖群體雜交組合方式。而通過全基因組關聯分析時,連鎖不平衡衰減越快,分析所需的標記密度越大,與目標性狀表型變異關聯的顯著性位點離功能基因位點距離會越近,進行全基因組關聯分析時,野生群體×養殖群體雜交組合方式可提高基因定位精度。
3.2 群體近交水平分析
水產動物具有較強的繁殖力,在短期內經過高強度的人工選擇可以實現性狀的快速改良,同時也意味著繁育的大部分個體父母本可能來自少數個體,育種群體的近交系數可能升高很快[14-15]。已有研究表明,貝類存在明顯的近交衰退問題[16-18]。了解群體的繁殖規律并掌握群體近交系數,有助于了解父母本的親緣關系,并通過雜交的方式降低近交的影響[12]。本研究結果顯示,野生群體、選育群體和養殖群體的群體內近交系數(Fis)分別為0.2075、0.3449和0.3465,選育群體和養殖群體接近近交系(Fis>0.375)[19]的水平,選育群體和養殖群體可以被視為具有一定純化水平的群系,無論是雜交育種或通過雜交方式降低群體近交系數,野生群體均可作為主要親本群。近交系數在進行雜交子代的遺傳參數分析時,可作為重要的參考依據。一般雜交父本和母本分別來自不同群體(系),父本和母本被默認為無親緣關系,但父本或母本個體間卻存在一定的親緣關系,利用基因型作為固定效應計算親緣系數會優于系譜計算。長純合片段反映了子代從有共同祖先的親代遺傳了相同的染色體片段[20],也可以幫助了解群體近交程度和群體多樣性,相較于群體內近交系數Fis,基于長純合片段所計算的近交系數FROH更接近于真實近交水平,已在畜禽遺傳研究中普遍應用[21-22],而本研究結果中,野生群體、選育群體和養殖群體的近交系數(FROH)分別為0.0149~0.0240、0.0159~0.0201和0.0170~0.0246,3個群體近交系數(FROH)相近并低于多數畜禽(FROH>0.05)[23-25],同時結果也明顯低于群體信息統計計算的近交系數,除物種差異因素外,結果偏低也與簡化測序結果與基因組覆蓋率低有關,且多數貝類沒有明顯性染色體[26],計算時以全基因組長度作為參數。近交系數(FROH)可作為群體內無直接親緣關系個體的親緣關系參考系數,在缺少基因型計算親緣系數時,近交系數(FROH)可直接作為合浦珠母貝選擇群體的近交系數進行因子計算,不同染色體長純合片段的分布也表明了3個群體從共同祖先遺傳染色體概率相近。
4 結 論
筆者通過分析廣西合浦珠母貝3個不同群體間的遺傳關系,發現不同群體間存在一定程度的遺傳分化,并指出通過野生群體×選育群體或野生群體×養殖群體組合方式開展雜交育種,具有較大獲得雜種優勢的潛力。研究結果可為廣西合浦珠母貝通過雜交育種對種質資源改良提供新的思路和途徑。
