白蛋白-奧曲肽納米顆粒制備及其緩釋性能
2023-03-20 11:48:32李昇原黃永鵬汪春江李秀蘭
中國粉體技術 2023年1期
關鍵詞:質量
李昇原, 黃永鵬, 唐 慧, 汪春江, 李秀蘭, 陳 博
(1. 四川輕化工大學 機械工程學院, 四川 宜賓 644000; 2. 國民核生化災害防護國家重點實驗室, 北京 102205)
多肽藥物藥理活性好,藥效專一性高,毒副作用小,在治療疾病時具有良好的應用前景[1-2],然而,多肽藥物存在易被酶降解、 半衰期短、 穩定性弱、 組織屏障通透性差等不足,造成在臨床應用上生物利用率低、 給藥頻繁、 患者依從性低等問題[3-5]。 納米緩釋制劑能夠提高藥物組織的滯留性和屏障穿透性,延長藥物的作用時間,進而改善多肽藥物存在的問題,是多肽藥物制劑研發的重點方向[6-8]。
人血清白蛋白(human serum albumin, HSA)是人體血漿中含量最豐富的蛋白,由585個氨基酸構成,半衰期約為19 d,具有安全無毒、 生物相容性好、 低免疫原性等優點,是理想的藥物載體[9-10]。人血清白蛋白納米顆粒(HSA-NPs)的制備方法有反溶劑法、 自組裝法、 乳化法、 凝膠法、 納米噴霧干燥法和納米白蛋白結合技術法等。反溶劑法通過向HSA溶液中添加反溶劑來降低HSA的溶解度,使HSA從水相脫溶、析出而獲得納米顆粒,工藝簡單,制備的顆粒粒徑較小,相較于其他方法具有明顯的優勢[11-13]。
將HSA制備成納米載體并與多肽藥物結合,能顯著提高多肽藥物在體內的穩定性及活性,避免過早被機體降解清除,延長體內的釋放與作用時間[14-16]。此外,臨床研究表明,HSA與奧曲肽(octreotide, OCT)等多肽藥物聯合用藥,能夠發揮更優的治療效果[17]。目前,以HSA為載體負載多肽藥物的納米緩釋藥劑研究尚處于起步階段。
本文中利用乙醇-水反溶劑法制備HSA-NPs,比較不同交聯劑的交聯性能,探討白蛋白質量濃度、 醇水體積比、 脫溶溫度、 溶劑pH、 交聯時間等工藝參數對HSA-NPs性能的影響,獲得制備HSA-NPs的優選工藝參數;選取OCT作為多肽模型藥物,通過浸漬吸附-冷凍干燥法構建HSA-OCT微粉制劑,初步探索制劑的藥物負載與釋放效應,為研發OCT及多肽藥物緩釋制劑提供研究基礎。
1 實驗
1.1 試劑材料
藥物:人血清白蛋白(HSA,質量分數為99%,上海吉至生化科技有限公司); 奧曲肽(OCT,質量分數為99.1%,批號為2018053003-1,浙江湃肽生物有限公司)。
交聯劑:戊二醛溶液(體積分數為50%,上海麥克林生化科技有限公司); 1, 6-己二異氰酸酯(體積分數為99%,北京百靈威公司); 1-乙基-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽(EDC)、 京尼平(質量分數為98%、 上海阿拉丁生化科技股份有限公司); 葡萄糖(分析純,北京化工廠)。
其他試劑: 無水乙醇、 氫氧化鈉、 鹽酸(分析純,北京化工廠); 乙腈、 高氯酸(色譜純,德國Merck公司); PBS緩沖液(pH為7.4)(武漢賽維爾生物科技有限公司)。
1.2 顆粒制備
1.2.1 白蛋白納米顆粒
稱取適量HSA, 溶于pH為3~11的水溶液中(用質量分數為0.1%的氫氧化鈉和鹽酸溶液調節), 配成質量濃度為5、 10、 20、 50 g/L的HSA溶液; 以轉速為500 r/min進行攪拌, 分別按醇水體積比為1∶1、 2∶1、 2.5∶1、 3∶1、 4∶1、 6∶1、 8∶1向HSA溶液中加入反溶劑無水乙醇; 待溶液出現藍色乳光, 加入交聯劑, 反應時間分別為3、 6、 12、 24 h, 最后旋轉蒸發除去乙醇, 得到膠體溶液; 將膠體溶液在轉速為18 000 r/min條件下離心處理20 min, 并用純水洗滌, 利用LGJ-18型真空冷凍干燥儀(北京松源華興科技公司)凍干得到HSA-NPs凍干粉。
1.2.2 白蛋白-奧曲肽微粉制劑
采用浸漬吸附-冷凍干燥法制備白蛋白-奧曲肽微粉制劑。取50 mg凍干后的HSA-NPs,加入30 mL質量濃度為3.33 g/L的OCT溶液, 在室溫條件下以轉速為500 r/min攪拌12 h, 得到載有OCT的HSA-NPs混懸液; 最后將混懸液進行離心處理除去上清液, 凍干后得HSA-OCT微粉制劑。
1.3 顆粒表征
利用BT-90+型動態光散射納米激光粒度儀(丹東百特儀器有限公司)測量HSA-NPs的粒徑和多分散指數(PDI), 對比不同時間點平均粒徑和PDI的變化情況, 研究納米顆粒的儲存穩定性; 采用ZS90型納米粒度電位儀(英國馬爾文儀器有限公司)測量HSA-NPs的Zeta電勢; 使用SU8020型掃描電鏡(日本Hitachi公司)觀察HSA-NPs凍干粉和HSA-OCT微粉制劑的微觀形貌。
1.4 白蛋白顆粒的綜合評價方法
運用Z分綜合評價法,從顆粒的性能、工藝穩定性2個方面篩選最優工藝條件。顆粒性能以平均粒徑小、 PDI值低為優;工藝穩定性用多次實驗所得平均粒徑和PDI值的相對標準偏差(RSD)值表示,RSD值越小工藝越穩定。將各指標的Z分等權相加得到Z分,Z分越大評價結果越優。單個指標的Z分的計算公式為
(1)
1.5 白蛋白-奧曲肽微粉制劑的評價方法
1.5.1 載藥量測定
采用1260型高效液相色譜法(美國Agilent公司)測定HSA-NPs中OCT的質量濃度。色譜柱Eclipse plus C18尺寸為φ4.6 mm×250 mm(直徑×長度), 色譜柱內的填料間孔徑為5 μm, 檢測波長為210 nm, 柱溫為25 ℃; 流動相中, 乙腈與質量分數為0.25%的高氯酸水溶液的體積比為30∶70,流量為1 mL/min,待檢測溶液的進樣體積為5 μL[18]。
取一定質量的HSA-OCT制劑置于離心管,加入純水溶解后超聲處理40 min并靜置一段時間,通過分析溶液中OCT的質量濃度測定載藥量。
1.5.2 藥物體外釋放性能的測定
藥物體外釋放性能參照藥物體外溶出度進行評定[19]。 取一定質量的HSA-OCT微粉制劑裝入透析袋, 加入PBS溶液至充盈透析袋, 保證藥物制劑完全潤濕; 扎緊透析袋兩端并浸沒在100 mL(溶出介質總體積)的PBS溶液中, 將裝有全部溶出介質容器置于磁力攪拌機上, 在37 ℃恒溫條件下以轉速為100 r/min進行攪拌。 按照設定的時間節點, 每次取出1 mL溶出介質后再補充1 mL新鮮介質, 分析溶出介質中OCT的質量濃度。 HSA-OCT微粉制劑的累計溶出度Q的計算公式為
(2)
式中:Ci和Cn分別為第i次和第n次最終取樣時OCT的質量濃度, g/L;m為OCT溶出總質量, mg;V1為每個時間節點取出的溶出介質體積, mL;V2為溶出介質總體積, mL。
2 結果與討論
2.1 交聯劑的選擇
HSA分子含有大量的極性基團,從水相脫溶析出的HSA-NPs不穩定,易復溶轉為水相或團聚成大顆粒[20-21]。交聯劑能連接HSA分子,穩定HSA-NPs。目前,HSA的交聯主要針對HSA游離的59個氨基[22]。
作為交聯劑的京尼平、 葡萄糖、 1, 6-己二異氰酸酯和戊二醛均能與HSA表面游離的氨基反應,交聯劑的加入質量為交聯HSA分子表面59個氨基理論所需質量的100%。在交聯過程中,EDC主要發揮活化HSA表面羧基、促使羧基與氨基偶聯的催化作用。結合文獻[23-24]和預實驗結果,EDC加入量為交聯HSA分子表面59個氨基理論所需量的10%,進一步提高EDC加入量會導致HSA分子過度交聯產生沉淀。
取1 mL質量濃度為10 g/L的HSA溶液, 攪拌狀態下加入2 mL無水乙醇, 交聯時間設為3 h, 不同交聯劑對HSA-NPs性能的影響如圖1所示。 由圖1(a)可見, 剛制備的HSA-NPs粒徑差距不大, 均小于200 nm; 儲存時間為10、 20 d以后, 未添加交聯劑或使用葡萄糖、 京尼平的HSA-NPs粒徑明顯增大, 平均粒徑大于750 nm, 添加交聯劑EDC、 1, 6-己二異氰酸酯、 戊二醛的HSA-NPs粒徑未有明顯變化。 由圖1(b)可見, 未添加交聯劑或使用葡萄糖、 京尼平作為交聯劑制備的顆粒的PDI值超過了0.25, 說明粒徑均一性較差, 添加交聯劑EDC、 1, 6-己二異氰酸酯、 戊二醛的HSA-NPs粒徑均一性較好。
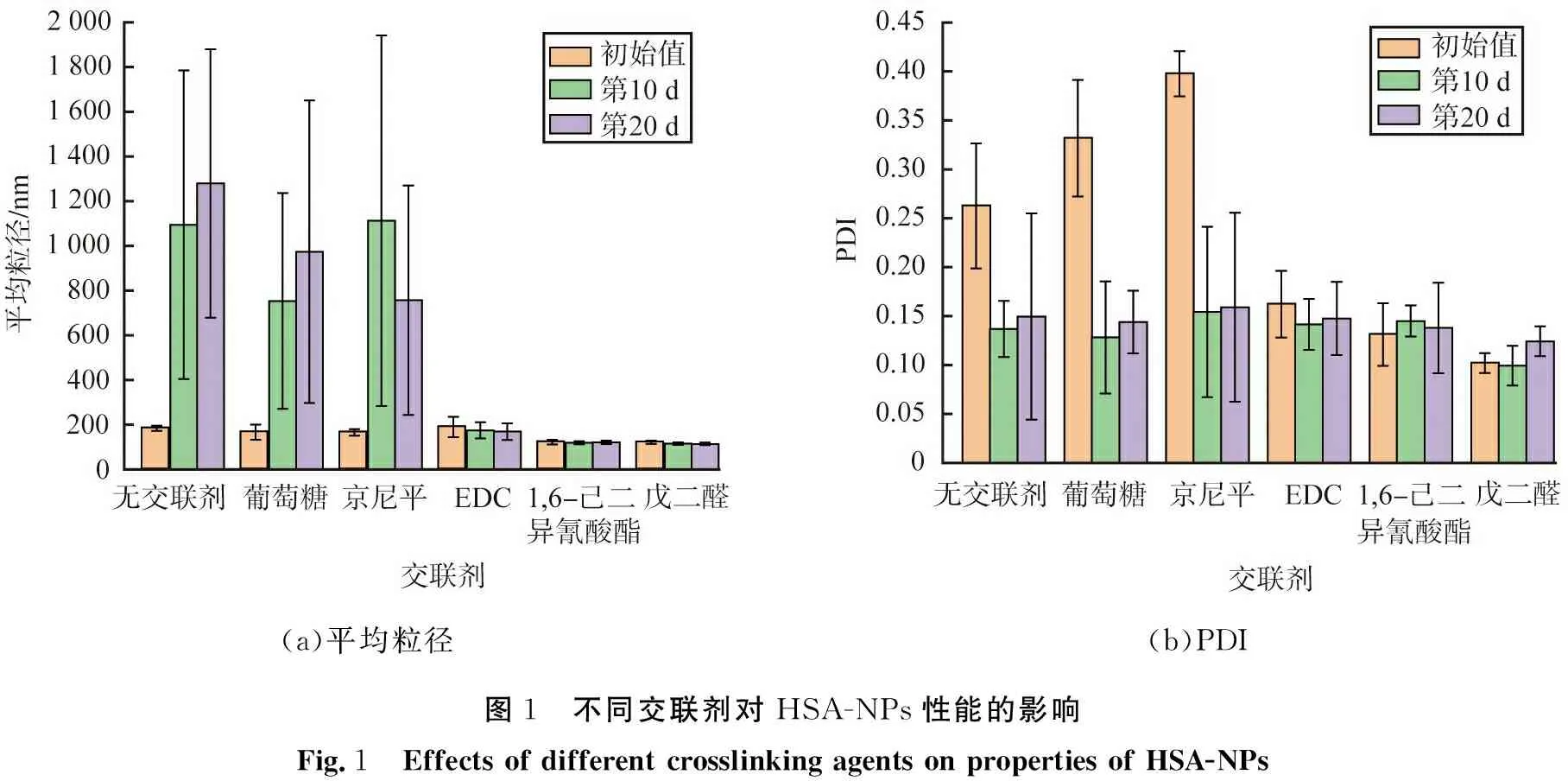
(a)平均粒徑(b)PDI圖1 不同交聯劑對HSA-NPs性能的影響Fig.1 Effects of different crosslinking agents on properties of HSA-NPs
儲存10 d后不同交聯劑制得的HSA-NPs的SEM圖像見圖2。由圖(a)—(c)可知,未添加交聯劑或使用葡萄糖、京尼平作為交聯劑制備的HSA-NPs顆粒之間有明顯得團聚現象;圖(e)、 (f)分別為使用1, 6-己二異氰酸酯和戊二醛制備的HSA-NPs顆粒,由圖可知納米顆粒呈圓球狀且具有較好的分散性; 圖(d)為EDC制備的HSA-NPs顆粒,對比發現顆粒之間存在一定程度的團聚,但團聚程度弱于未添加交聯劑或使用葡萄糖、 京尼平作所制備的顆粒。

(a)無交聯劑(b)葡萄糖(c)京尼平(d)EDC(e)1, 6-己二異氰酸酯(f)戊二醛圖2 儲存10 d后不同交聯劑制得的HSA-NPs的SEM圖像Fig.2 SEM images of HSA-NPs prepared with different crosslinking agents after storage for 10 days
反溶劑法制備HSA-NPs的實質是HSA分子從水相均相成核、 長大的過程。由于納米顆粒表面能的增大,因此該過程還伴隨著顆粒的團聚現象。交聯劑不改變HSA溶液的過飽和度,因此,顆粒的粒徑及穩定性由團聚傾向決定。HSA的等電點為4.8[25],當HSA處于非等點狀態時,其表面會附帶同種電荷。同種電荷數量越多,顆粒之間排斥力越強,就越能抵消顆粒團聚傾向,使顆粒保持穩定。交聯劑能通過改變納米顆粒表面的電荷量影響顆粒的粒徑、 分散性和穩定性。納米顆粒均帶負電荷,不同交聯劑制得的HSA-NPs的Zeta電勢絕對值如圖3所示。由圖可見,未交聯或使用葡萄糖、 京尼平交聯后HSA-NPs的Zeta電勢較小,顆粒附帶的電荷量較少,顆粒間排斥力弱,因此不穩定,易團聚;因為不穩定的顆粒易在交聯劑的作用下形成更大的顆粒,使顆粒粒徑分布更寬,所以葡萄糖、 京尼平交聯后的顆粒PDI值更大,粒徑均一性更差;1, 6-己二異氰酸酯和戊二醛交聯的納米顆粒Zeta電勢較大,顆粒具有較多的同種電荷,排斥力較強,沒有團聚現象,制備的HSA-NPs性能較好;EDC交聯的納米顆粒Zeta電勢處于中間狀態,顆粒的電荷量介于二者之間,制備的HSA-NPs存在一定程度的團聚現象。
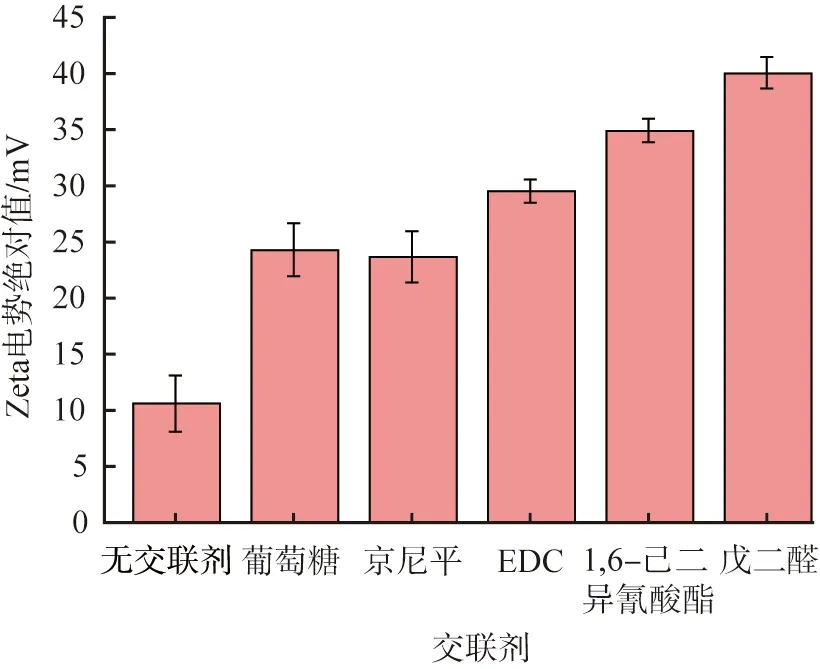
圖3 不同交聯劑制得的HSA-NPs的Zeta電勢Fig.3 Zeta potential of HSA-NPs prepared with different crosslinking agents
表1為不同交聯劑制得的HSA-NPs的Z分評價結果。由表可以看出,EDC、 戊二醛、 1, 6-己二異氰酸酯作為交聯劑制備的HSA-NPs的平均粒徑分別為(190.0±19.45)、 (121.7±4.70)、 (122.9±7.1) nm, PDI值分別為0.13±17.18、 0.13±8.14、 0.13±19.85;Z分分別為-4.34、 -4.34、 0.40;戊二醛交聯的HSA-NPs納米顆粒的平均粒徑、 PDI值和工藝穩定性均較好,說明Z分最大的戊二醛為優選交聯劑。葡萄糖、 京尼平的交聯能力較差,不納入評價。
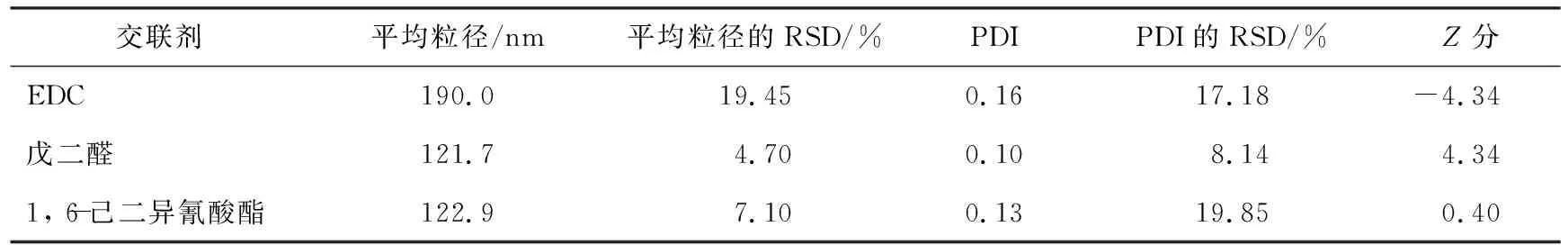
表1 不同交聯劑制得的HSA-NPs的Z分評價結果
2.2 白蛋白納米顆粒的優選工藝參數
2.2.1 白蛋白質量濃度
以戊二醛作為交聯劑, 設定醇水體積比為2∶1, 交聯時間為3 h, pH為7, 脫溶溫度為20 ℃, HSA質量濃度對HSA-NPs性能的影響如圖4所示。 由圖可知, 隨著HSA質量濃度的增加, HSA-NPs納米顆粒的粒徑逐漸減小, PDI值先減小后增大, 各項工藝參數的Z分則先增大后減小; 當HSA的質量濃度為10 g/L時,Z分最大為0.88,因此,根據Z分評價結果, HSA的優選質量濃度為10 g/L。

圖4 HSA的質量濃度對HSA-NPs性能的影響Fig.4 Effect of HSA mass concentration on HSA-NPs performance
根據液相均勻成核理論,顆粒的成核、 生長速率是影響粒徑及其分布的主要因素。質量濃度大的HSA溶液具有較大的過飽和度,顆粒成核、 生長速率快,所得納米顆粒小,因此HSA-NPs的平均粒徑隨HSA質量濃度的增大而逐漸減小;另一方面,質量濃度大的HSA溶液生成的顆粒較多,顆粒之間更易碰撞、 團聚,同時由于溶液的黏度較大,降低了HSA分子在水和乙醇之間的擴散速率,因此HSA-NPs的粒徑分布較寬,PDI值較大。對于質量濃度較小的HSA溶液,顆粒成核、 生長速率較慢,所需脫溶反應時間較長,使得納米顆粒的粒徑分布同樣不均勻,適中的成核速率能保證顆粒的粒徑及其分布均較優。
當傳統的題材進入到陶瓷的領域中也在陶瓷領域的疆土中開辟出獨具花鳥瓷畫風格的一片疆土。這種與眾不同的瓷畫表現形式讓皇族到百姓,無一不為之美感而折服,從而使花鳥瓷畫享譽世界。
2.2.2 醇水體積比
以戊二醛作為交聯劑, 設定交聯時間為3 h, pH為7, 脫溶溫度為20 ℃, HSA質量濃度為10 g/L, 醇水體積比對HSA-NPs性能的影響如圖5所示。 由圖可以看出, 當醇水體積比為1∶1時納米顆粒平均粒徑為(267.56±11.3) nm, PDI值為0.19±21.8; 醇水體積比為2∶1、 2.5∶1、 8∶1時,能獲得較小的平均粒徑,分別為(102.9±7.9)、 (86.9±24.6)、 (107.7±15.25) nm, 醇水體積比為2.5∶1、 8∶1時制備的納米顆粒的PDI值大于0.25。 醇水體積比為3∶1、 4∶1、 6∶1時平均粒徑超過了1 200 nm, 顆粒的性能較差, 不納入Z分計算范圍。 當醇水體積比為2∶1時,Z分最大為1.54, 因此, 根據Z分評價結果, HSA-NPs的優選醇水體積比為2∶1。

圖5 醇水體積比對HSA-NPs性能的影響Fig.5 Effects of volumn ratio of alcohol to water on HSA-NPs performance
醇水體積比會改變溶液的過飽和度, 從而影響體系內顆粒的粒徑、 分布情況, 不同醇水體積比制備的HSA-NPs粒徑分布如圖6所示。 由圖可見, 醇水體積比為1∶1時, 溶液產生的過飽和度較低, 顆粒成核、 生長速率慢, 制備的顆粒粒徑為100~500 nm,由于溶劑體積小,顆粒間易碰撞發生團聚,因此體系中還存在大于1 000 nm的大顆粒;醇水體積比為2∶1時,過飽和度有明顯提高,此時生成的納米顆粒粒徑為50~250 nm,粒徑分布較為集中;隨著醇水體積比進一步的增大,溶液產生的過飽和度逐漸增大,能生成更細小的粒徑小于50 nm顆粒,但小顆粒團聚傾向大,使得顆粒呈多種分散狀態;醇水體積比為2.5∶1時,開始存在明顯的團聚現象,制備的顆粒粒徑為15~700 nm,因為體系內小顆粒數量較多,所以HSA-NPs的平均粒徑小,但粒徑分布寬,PDI值大;醇水體積比達到4∶1時,顆粒的團聚傾向最大,體系存在大量的大顆粒;繼續增加醇水體積比,溶液中產生的過飽和度趨于恒定,但溶劑體積逐漸增大,降低了顆粒碰撞、 團聚的概率,因此顆粒的粒徑有所減小;當醇水體積比為8∶1時,團聚傾向明顯減弱,體系中只存在少量大顆粒。綜上,從粒徑分布情況來看,當醇水體積比為2∶1時,制備的HSA-NPs顆粒的性能最優。
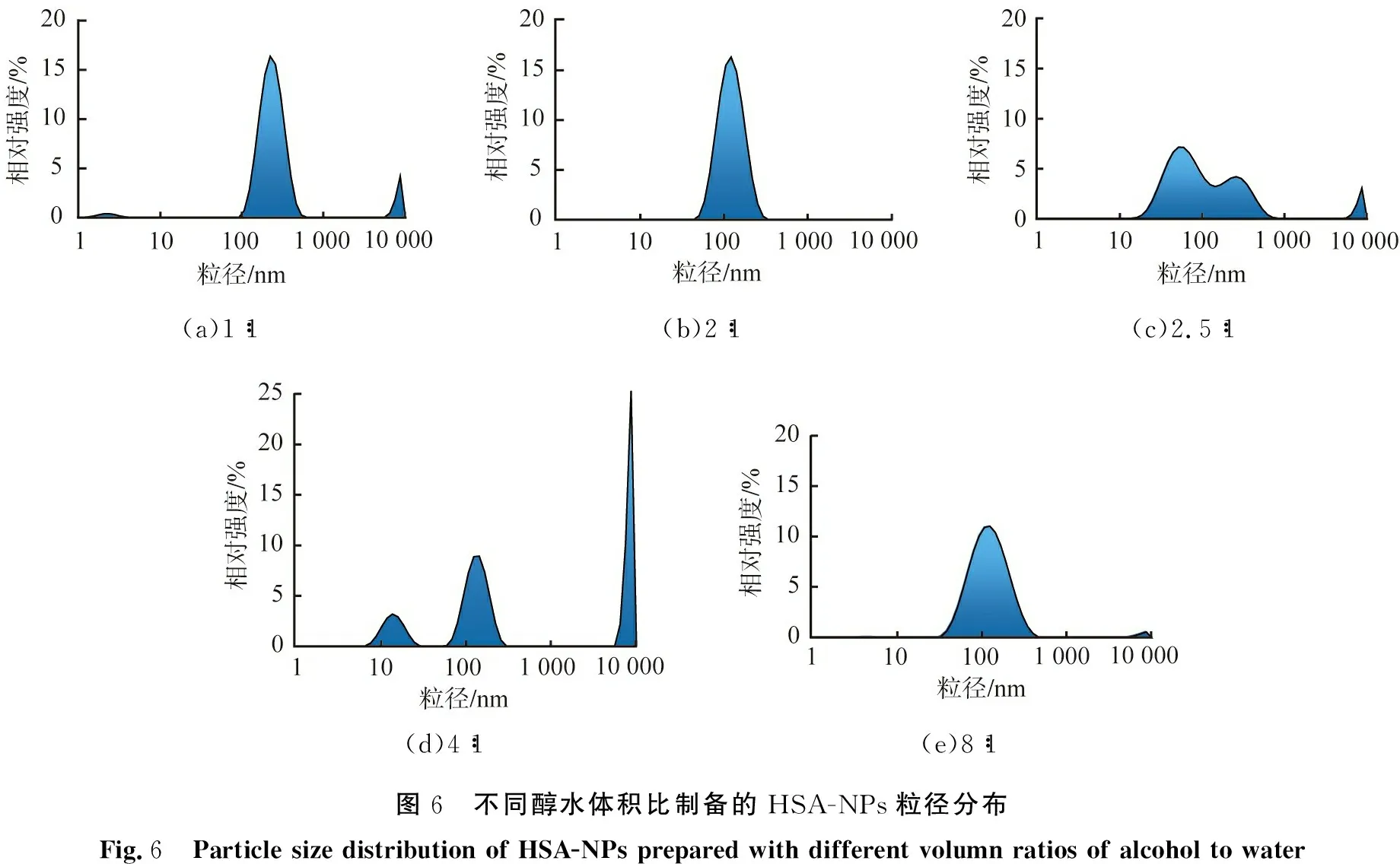
(a)1∶1(b)2∶1(c)2.5∶1(d)4∶1(e)8∶1圖6 不同醇水體積比制備的HSA-NPs粒徑分布Fig.6 Particle size distribution of HSA-NPs prepared with different volumn ratios of alcohol to water
2.2.3 脫溶溫度
以戊二醛作為交聯劑,設定交聯時間為3 h,pH為7,HSA質量濃度為10 g/L,醇水體積比為2∶1,將HSA溶液進行水浴,脫溶溫度對納米顆粒性能的影響如圖7所示。由圖可見,當脫溶溫度為5 ℃時,納米顆粒的平均粒徑與PDI值均較大;當脫溶溫度為10~30 ℃時,隨溫度的升高,HSA-NPs的平均粒徑逐漸增大,PDI值逐漸減小;Z分隨著脫溶溫度的升高呈現先增大后減小的變化規律;當脫溶溫度為20 ℃時,Z分最大為2.68,因此,根據Z分評價結果,HSA-NPs的優選脫溶溫度為20 ℃。

圖7 溫度對HSA-NPs性能的影響Fig.7 Effects of temperature on HSA-NPs performance
實驗發現,脫溶溫度為5 ℃時,溶液未觀察到明顯的藍色乳光,表明體系內生成的納米顆粒數量較少,低溫抑制了HSA分子脫溶析出過程。當脫溶溫度為10~30 ℃時,HSA分子能正常脫溶析出生成納米顆粒,一方面,隨著脫溶溫度的升高,溶液產生的飽和度逐漸降低,體系內顆粒成核與生長速率減緩,使得制備的顆粒平均粒徑逐漸增大,溫度通過改變HSA的溶解度而直接影響過飽和度;另一方面,溫度會影響分子的擴散速率,溫度越高分子擴散速率越快,微觀混合越充分,制備的顆粒粒徑分布就均勻。綜上,從溫度對HSA的過飽和度和擴散速率的影響來看,優選脫溶溫度為20 ℃。
2.2.4 溶劑pH

圖8 溶劑pH對HSA-NPs性能的影響Fig.8 Effects of solvent pH on HSA-NPs performance
因為HSA分子在不同pH溶液中帶電量不同,所以可以通過調節溶劑pH來改變HSA分子的靜電作用, 制備不同粒徑的納米顆粒。然而, HSA分子中的咪唑環(位于16個組氨酸上)以及末端的氨基和羧基能夠被質子化, 使其具有一定的酸堿緩沖能力[26], 同時, 調節溶劑pH時引入的離子, 會削弱納米顆粒之間的靜電排斥力[27], 使粒徑略大于中性溶液中的顆粒。
2.2.5 交聯時間
以戊二醛作為交聯劑,設定HSA質量濃度為10 g/L,醇水體積比為2∶1,脫溶溫度為20 ℃時, 溶劑pH為9時, 交聯時間對HSA-NPs性能的影響如圖9所示。 由圖可見, 交聯時間對顆粒的平均粒徑和PDI值影響不大; 不同交聯時間下, 顆粒的平均粒徑均小于120 nm, 分散性良好。 當交聯時間為24 h時,Z分最大為1.52, 因此, 根據Z分評價結果, HSA的優選交聯時間為24 h。
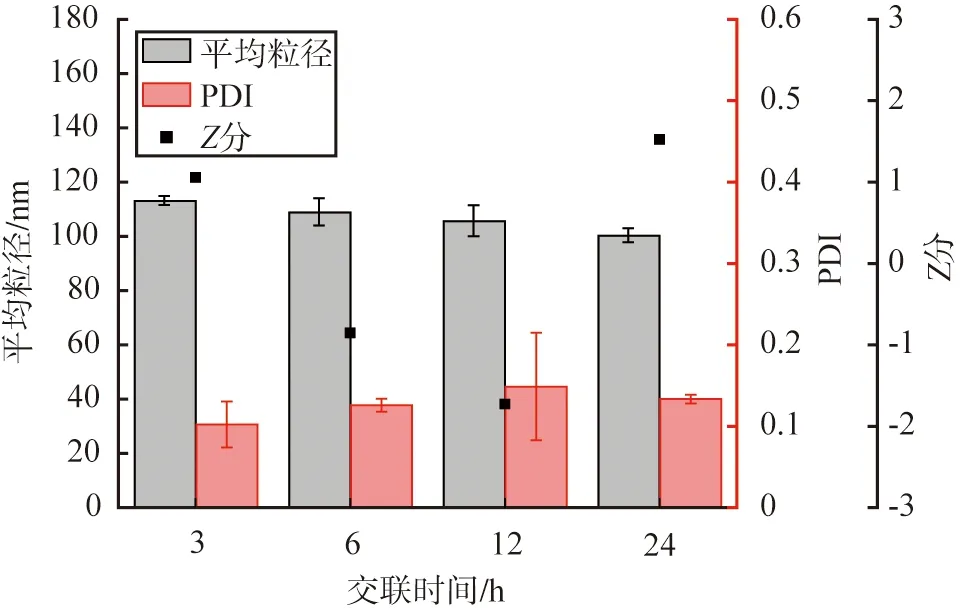
圖9 交聯時間對HSA-NPs性能的影響Fig.9 Effects of crosslinking time on HSA-NPs performance
2.2.6 優選工藝條件
綜合上述評價結果, 制備HSA-NPs的優選工藝參數為: HSA的質量濃度為10 g/L; 醇水體積比為2∶1; 脫溶溫度為20 ℃;溶劑pH為9;交聯時間為24 h。在此條件下制備的HSA-NPs工藝穩定性良好,平均粒徑為(100.6±2.5) nm, PDI為0.13±0.01。
2.3 納米顆粒的載藥量與體外釋放性能
HSA-NPs凍干粉與HSA-OCT制劑的SEM圖像如圖10所示。由圖可以看出,載藥前、 后納米顆粒的形態未發生明顯變化,保持為規則的球狀形貌,經計算,HSA-OCT微粉制劑的載藥量為1.83%。對載藥機理進行分析后發現,OCT的等電點為8.2[28],在純水中OCT分子表面帶正電荷,因此能與帶負電荷的HSA-NPs發生靜電作用,吸附于納米顆粒表面;同時HSA分子中的α-螺旋形成了較多的網狀空隙,為OCT的吸附提供了有利空間。

(a)HSA-NPs凍干粉(b)HSA-OCT制劑圖10 HSA-NPs凍干粉與HSA-OCT制劑的SEM圖像Fig.10 SEM images of HSA-NPs freeze-dried powder and HSA-OCT preparation
HSA-OCT制劑和OCT原藥的累計溶出度隨時間的變化曲線如圖11。由圖可見,OCT原藥在累計釋放時間為120 min內時累計溶出度達到了90.6%,累計釋放時間為480 min后完全溶出;HSA-OCT制劑較OCT原藥釋放緩慢,有較明顯的緩釋作用, 藥物累計釋放時間達到2 160 min, 但時間為0~30 min時卻無藥物溶出, 可能是因為HSA-NPs的吸附能力較強, 需要經過一定時間后OCT才能釋放。

(a)0~2 500 min(b)0~60 min圖11 HSA-OCT制劑和OCT原藥的累計溶出度隨時間的變化曲線Fig.11 Curve of cumulative dissolution of HSA-OCT preparation and OCT agent changing with time
3 結論
本文中采用反溶劑法制備HSA-NPs,對比了不同交聯劑的交聯性能;研究了白蛋白質量濃度、 醇水體積比、 脫溶溫度、 溶劑pH、 交聯時間等工藝參數對HSA-NPs性能的影響;運用浸漬吸附-冷凍干燥法構建了HSA-OCT微粉制劑,分析了OCT原藥和HSA-OCT微粉制劑的體外緩釋性能。
1)交聯劑能改變HSA分子表面的靜電作用,降低HSA-NPs的團聚傾向,從而穩定固化HSA-NPs。在所選交聯劑中,戊二醛交聯的HSA-NPs納米顆粒的性能、工藝穩定性最優,制備的顆粒微觀形貌為規則球狀,儲存穩定性良好。經Z分綜合評價戊二醛為優選交聯劑。
2)HSA質量濃度、 醇水體積比和脫溶溫度對HSA-NPs的性能影響較大。 根據Z分評價結果, 制備HSA-NPs顆粒的優選工藝參數為: HSA質量濃度為10 g/L, 醇水體積比為2∶1, 脫溶溫度為20 ℃, 溶劑pH為9, 交聯時間為24 h。 在優選工藝參數參數條件下制備的納米顆粒的平均粒徑為(100.6±2.5) nm, PDI為0.13±0.01。
3)載藥前后HSA-NPs微觀形貌未發生明顯變化。相較于OCT原藥,HSA-OCT微粉制劑具有明顯的緩釋效應,持續釋藥時間為2 160 min,具有較好的體外釋放性能。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
