阿司匹林丁香酚酯對肝臟主要藥物代謝酶活性的影響
2023-03-08 03:12:36郝若晨劉希望李世宏白莉霞李劍勇楊亞軍
中國獸醫(yī)學(xué)報 2023年2期
關(guān)鍵詞:小鼠
陶 琦,郝若晨,劉希望,秦 哲,李世宏,白莉霞,李劍勇,楊亞軍
(中國農(nóng)業(yè)科學(xué)院 蘭州畜牧與獸藥研究所 農(nóng)業(yè)農(nóng)村部獸用藥物創(chuàng)制重點實驗室/甘肅省新獸藥工程重點實驗室,甘肅 蘭州 730050)
藥物進(jìn)入體內(nèi)后,有些藥物能夠提高肝臟微粒體藥物代謝酶系(藥酶)的活性,從而提高藥物代謝速率,稱為酶的誘導(dǎo);或者使藥酶的活性降低,使其代謝藥物的速率減慢,稱為酶的抑制。藥酶的誘導(dǎo)或抑制均可影響藥物代謝的速率;當(dāng)2種或2種以上的藥物在同時或前后序貫用藥時,如果在代謝環(huán)節(jié)產(chǎn)生了藥酶的誘導(dǎo)或抑制,使療效增強(qiáng)甚至產(chǎn)生毒副作用,或療效減弱甚至治療失敗,此稱之為代謝性藥物相互作用(metabolic drug interaction)[1]。隨著藥物聯(lián)合使用越來越普遍,藥物之間的代謝性相互作用愈顯重要。在臨床用藥中,相當(dāng)部分的藥物不良反應(yīng)是由代謝性藥物相互作用引起的,其發(fā)生率占藥代動力學(xué)相互作用的40%,而代謝性相互作用的96%是由肝臟微粒體細(xì)胞色素P450酶系統(tǒng)(cytochrome P450,CYP450)介導(dǎo)的[2-3]。研究藥物對CYP450酶系的影響,可以預(yù)測其在體內(nèi)可能引起的代謝性藥物相互作用,進(jìn)而為臨床藥物相互作用試驗提供參考。因此,在新藥的安全性評價中,研究藥物對CYP450酶的活性影響具有重要意義。
目前,歐美各國已經(jīng)將藥物對CYP450酶活性影響的測定用于代謝及新藥的篩選研究,并將其列為新藥研究中必須進(jìn)行的一項試驗[4-6]。我國于2021年頒布了《藥物相互作用研究指導(dǎo)原則(試行)》,明確了化學(xué)藥物相互作用研究的具體方法,生物制品、中藥和天然藥物可參照執(zhí)行[7]。
阿司匹林丁香酚酯(aspirin eugenol ester,AEE)是利用結(jié)構(gòu)拼合的前藥原理,以丁香酚(Eug)對阿司匹林(Asp)進(jìn)行酯化修飾而合成的一種新型藥用化合物,具有很好的成藥性。研究表明,AEE具有確切的抗炎[8]、抗氧化[9-10]、降血脂[11-12]、預(yù)防動脈粥樣硬化[11-12]、預(yù)防血栓形成[15-17]和抗急性肝損傷作用[18]。前期研究主要集中在藥理與毒理方面,該藥物對CYP450酶活性的影響尚不清楚。因此,本試驗選擇SD大鼠和C57小鼠為研究對象,將其分為空白對照組與試驗組,采用ELISA方法,考察AEE對CYP450酶的6種主要亞型CYP3A4、CYP2C8、CYP2C19、CYP1A2、CYP2D6和CYP2C9活性的影響,預(yù)測AEE在動物體內(nèi)可能引起的代謝性藥物相互作用,為研究AEE與其他藥物的相互作用及其作用機(jī)制奠定基礎(chǔ),為其獸醫(yī)臨床試驗研究提供參考依據(jù)。
1 材料與方法
1.1 藥品與試劑AEE(含量99.8%,中國農(nóng)業(yè)科學(xué)院蘭州畜牧與獸藥研究所自制,批號:20190815);羧甲基纖維素鈉,大鼠CYP1A2試劑盒(批號:ml592615-J),大鼠細(xì)胞色素氧化酶2C8(CYP2C8)試劑盒(批號:ml696555-J),大鼠細(xì)胞色素P450家族成員2C9(CYP2C9)試劑盒(批號:ml846955-J),大鼠細(xì)胞色素P450家族成員2C19(CYP2C19)試劑盒(批號:ml565952-J),大鼠細(xì)胞色素P450酶亞型2D6(CYP2D6)試劑盒(批號:ml986556-J),大鼠細(xì)胞色素P450家族成員3A4(CYP3A4)試劑盒(批號:ml365254-J),小鼠CYP1A2試劑盒(批號:ml875116-J),小鼠細(xì)胞色素P450氧化酶2C8(CYP2C8)試劑盒(批號:ml763516-J),小鼠細(xì)胞色素P450家族成員2C9(CYP2C9)試劑盒(批號:ml563432-J),小鼠細(xì)胞色素P450家族成員2C19(CYP2C19)試劑盒(批號:ml578931-J),小鼠細(xì)胞色素P450酶亞型2D6(CYP2D6)試劑盒(批號:ml963546-J),小鼠細(xì)胞色素P450家族成員3A4(CYP3A4)試劑盒(批號:ml568741-J),以上試劑盒均購自上海酶聯(lián)生物科技有限公司;磷酸鹽緩沖液(PBS)(索萊寶,批號:D1040-500);烏拉坦(NJKOCED);蛋白酶抑制劑(Thermo Fisher,貨號:36978)。
1.2 實驗動物40只清潔級SD大鼠,雄性,1月齡,體質(zhì)量110~120 g;40只清潔級C57小鼠,雄性,6~8周齡,體質(zhì)量18~22 g,均購自中國農(nóng)業(yè)科學(xué)院蘭州獸醫(yī)研究所,動物生產(chǎn)許可證號:NKMYD201907018。
1.3 溶液配制稱取羧甲基纖維素鈉適量,用蒸餾水配制成0.5%的溶液。試驗前將AEE用研缽研磨成細(xì)粉。將研磨的AEE混懸在 0.5%的羧甲基纖維素鈉中,配制成質(zhì)量濃度為20 g/L的混懸液備用。
1.4 試驗分組及給藥方式將SD大鼠及C57小鼠,分別隨機(jī)分為空白對照組和試驗組,每組20只。空白組灌服0.5%的羧甲基纖維素鈉溶液;試驗組灌服AEE(54 mg/kg)。每天1次,連續(xù)給藥3周后,每組隨機(jī)選取10只,禁食不禁水過夜,腹腔注射烏拉坦麻醉后,迅速處死,取出肝臟,用預(yù)冷的PBS(4℃預(yù)冷2 h)沖洗組織表面,去除表面殘留的血液。各組的剩余大鼠,于停藥2周后,以相同方式采集肝臟。
1.5 樣品采集與處理將肝臟樣品測質(zhì)量后剪碎,與PBS(PBS中加入了蛋白酶抑制劑)按比例(1 g∶9 mL)混合,置于勻漿機(jī)中充分均質(zhì)后,進(jìn)行超聲破碎,以使進(jìn)一步裂解,然后4℃、5 000×g離心10 min,取上清液備用。上述操作均在冰浴條件下進(jìn)行。
1.6 肝臟藥物代謝酶活性的測定設(shè)置標(biāo)準(zhǔn)品孔和樣品孔。根據(jù)試劑盒說明書操作步驟進(jìn)行測定,在450 nm波長處測定各孔的D值。以所測標(biāo)準(zhǔn)品的D值為縱坐標(biāo),標(biāo)準(zhǔn)品的濃度值為橫坐標(biāo),繪制標(biāo)準(zhǔn)曲線,并得到直線回歸方程,將樣品的D值代入方程,計算出樣品中酶的活性。
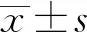
2 結(jié)果
2.1 SD大鼠與C57小鼠體質(zhì)量變化受試動物適應(yīng)一段時間后,開始正式試驗。定期對其體質(zhì)量進(jìn)行記錄。如圖1所示,2組SD大鼠的體質(zhì)量在給藥和停藥期間無顯著性差異(P>0.05)。如圖2所示,2組C57小鼠的體質(zhì)量在給藥和停藥期間無顯著性差異(P>0.05)。

圖2 C57小鼠隨時間體質(zhì)量變化
2.2 標(biāo)準(zhǔn)曲線的建立根據(jù)試劑盒說明書分別建立SD大鼠和C57小鼠肝臟藥物代謝酶標(biāo)準(zhǔn)曲線。結(jié)果顯示,SD大鼠和C57小鼠CYP450酶的6種主要亞型CYP3A4、CYP2C8、CYP2C19、CYP1A2、CYP2D6、CYP2C9的標(biāo)準(zhǔn)曲線的相關(guān)系數(shù)R2均在0.99以上(圖3,4)。說明在各質(zhì)量濃度范圍內(nèi),各酶標(biāo)準(zhǔn)曲線的線性良好,可以進(jìn)行下一步試驗。

A~F.分別為CYP3A4、CYP2C8、CYP2C19、CYP1A2、CYP2D6、CYP2C9亞型

A~F.分別為CYP3A4、CYP2C8、CYP2C19、CYP1A2、CYP2D6、CYP2C9亞型
2.3 給藥后肝臟藥物代謝酶活性如圖5所示,SD大鼠連續(xù)灌服給藥3周后,與對照組比較,AEE對CYP3A4、CYP2C19、CYP1A2的活性有顯著的抑制作用(P<0.05),對CYP2C8的活性有顯著的誘導(dǎo)作用(P<0.05),而對CYP2D6、CYP2C9的活性無顯著影響(P>0.05)。

A~F.分別為CYP3A4、CYP2C8、CYP2C19、CYP1A2、CYP2D6、CYP2C9亞型。與空白對照組相比*P<0.05。下同
如圖6所示,C57小鼠在連續(xù)灌藥3周后,其酶活性結(jié)果與SD大鼠相似,即對CYP3A4、CYP2C19、CYP1A2、CYP2D6的活性有顯著抑制作用(P<0.05),對CYP2C8的活性有一定的促進(jìn)作用(P<0.05),對CYP2C9無顯著影響(P>0.05);然而,AEE給藥后,C57小鼠CYP2D6的活性亦出現(xiàn)了明顯的抑制(P<0.05)。

A~F.分別為CYP3A4、CYP2C8、CYP2C19、CYP1A2、CYP2D6、CYP2C9亞型
2.4 停藥后肝臟藥物代謝酶活性如圖7所示,SD大鼠停藥2周后,與對照組比較,僅CYP2C19的活性尚未恢復(fù),而其余藥酶活性皆有一定的恢復(fù),與對照組無顯著性差異(P>0.05)。

A~F.分別為CYP3A4、CYP2C8、CYP2C19、CYP1A2、CYP2D6、CYP2C9亞型
如圖8所示,C57小鼠停藥2周后,與對照組比較,發(fā)現(xiàn)只有CYP2C19的活性沒有恢復(fù),而其余藥酶活性都有一定的恢復(fù),與對照組無顯著性差異(P>0.05)。

A~F.分別為CYP3A4、CYP2C8、CYP2C19、CYP1A2、CYP2D6、CYP2C9亞型
2.5 AEE對2種實驗用鼠的肝臟主要藥物代謝酶活性的影響AEE試驗組與空白對照組2組實驗鼠,在試驗期間體質(zhì)量無顯著性差異(P>0.05),說明AEE在本試驗期間對實驗鼠體質(zhì)量無顯著性影響。本試驗主要考察了AEE對CYP450酶的6種亞型:CYP3A4、CYP2C8、CYP2C19、CYP1A2、CYP2D6、CYP2C9的影響以及影響后的自我恢復(fù)。結(jié)果顯示,AEE對CYP3A4、CYP2C19、CYP1A2的活性有一定的抑制作用(P<0.05),對CYP2C8的活性有一定的誘導(dǎo)作用(P<0.05),而CYP2C9的活性無顯著性差異(P>0.05);停藥2周后,發(fā)現(xiàn)只有CYP2C19的活性沒有恢復(fù),而其余藥酶活性都有一定的恢復(fù),與空白對照組無顯著性差異(P>0.05),詳見表1。

表1 藥酶(CYP)活性變化
3 討論
CYP450酶屬于血紅素蛋白基因超家族,編碼一系列的代謝酶,主要存在于肝臟內(nèi)質(zhì)網(wǎng)上[18]。該酶參與了大多數(shù)內(nèi)源性和外源性化合物的代謝,藥物的代謝速率依賴于其活性。已知的CYP450中主要有CYP1、CYP2、CYP3 三個家族,參與機(jī)體內(nèi)的內(nèi)源性和外源性化合物的生物轉(zhuǎn)化[20]。CYP1A2是CYP450酶超家族中CYP1的重要亞型,主要分布在肝臟,參與約5%的臨床藥物,如咖啡因、非那西丁、茶堿、氯氮平和三環(huán)類抗抑郁藥的代謝過程,同時在前致癌物與前毒物的生物轉(zhuǎn)化過程中也起到了重要的作用[21]。CYP2C這個家族是CYP450超家族中最大的亞家族,包括CYP2C8、CYP2C9、CYP2C10、CYP2C19等多個亞型[22-24]。CYP2D6是一種主要的藥物代謝酶,盡管只占肝臟酶總量的2%左右,但負(fù)責(zé)消除超過20%~30%的臨床使用藥物[25]。
CYP450酶的活性可被外源化合物影響,呈現(xiàn)可誘導(dǎo)性或可抑制性,從而在聯(lián)合用藥的過程中產(chǎn)生藥物-藥物相互作用,使得藥效降低或發(fā)生藥物不良反應(yīng)[26]。一些CYP450可被誘導(dǎo),而有些卻對此不敏感;前者稱為誘導(dǎo)性P450,后者稱為構(gòu)成性P450[27]。有誘導(dǎo)作用的化合物大多數(shù)是該酶的底物或類似物[28],在生物合成中的作用是催化。化合物對CYP450酶的抑制可分為4類:可逆性競爭抑制、可逆性非競爭抑制、不可逆性抑制、干擾蛋白質(zhì)合成。可逆性競爭抑制:同一種酶的兩種底物競爭相同活性部位互為抑制,這是由底物重疊性的特點所決定的。如,美托洛爾和異喹胍都是CYP2D6的底物,當(dāng)它們同時存在時,就存在競爭抑制[29]。可逆性非競爭抑制過程:物質(zhì)本身可能無抑制作用,但其代謝產(chǎn)物與相應(yīng)酶的親和力較高,從而抑制物質(zhì)本身的進(jìn)一步代謝。隨著代謝產(chǎn)物的消除,這種抑制作用也隨之消除。如,食用調(diào)味品的成分黃樟醚的代謝過程[30]。不可逆性抑制:P450結(jié)構(gòu)被破壞或造成蛋白質(zhì)永久失活。如,農(nóng)藥1605、馬拉硫磷對P450的抑制作用[30]。干擾蛋白質(zhì)合成:動物機(jī)體若營養(yǎng)不良或有肝臟疾病,可抑制蛋白質(zhì)P450的合成,從而抑制P450的活性。研究藥物對CYP450酶的誘導(dǎo)和抑制,對于臨床合理用藥、提高藥物療效、降低藥物的毒副作用具有重要意義。因此,本試驗采用ELISA方法考察AEE對2種實驗鼠的肝臟主要藥物代謝酶活性的影響。
CYP2D6是CYP450酶系中重要的一種氧化代謝酶,其基因多態(tài)性對酶的活性有著至關(guān)重要的影響,是構(gòu)成藥物代謝個體差異和種族差異的基礎(chǔ)[31-32],CYP2D6基因多態(tài)性對藥物代謝起著重要的作用。本試驗中AEE對SD大鼠和C57小鼠的CYP2D6活性的影響存在差異,原因可能是由于2種實驗鼠的CYP2D6的基因型有差異,從而導(dǎo)致AEE對2種實驗鼠的CYP2D6活性影響有一定的差異。
CYP3A4是CYP450酶中最重要的成分,約占CYP450酶總量的30%~40%,該酶在許多內(nèi)外源化合物的代謝中起著重要作用。本試驗結(jié)果表明AEE能夠抑制CYP3A4的活性,研究發(fā)現(xiàn)阿司匹林也能夠抑制CYP3A4的活性[33-34],提示AEE作為阿司匹林的衍生物,作用效果可能與阿司匹林相似。
CYP2C19是S-MP4羥化酶(S-美芬妥英羥化酶)[35],是第一代質(zhì)子泵抑制劑奧美拉唑、蘭索拉唑、泮托拉唑等藥物的最主要代謝酶,其中奧美拉唑90%都是通過CYP2C19進(jìn)行代謝的[36]。而AEE對CYP2C19的活性具有抑制作用,提示AEE與奧美拉唑聯(lián)用,可能會提高奧美拉唑?qū)ο罎兊闹委熜Ч?/p>
CYP1A2是CYP450酶中CYP1的重要亞型,主要分布在肝臟,參與約5%臨床藥物代謝。其活性與藥物的療效或毒性以及某些腫瘤的易感性密切相關(guān),具有重要的生理學(xué)和毒理學(xué)意義[37-38]。AEE對CYP1A2的活性具有抑制作用,其作用機(jī)制需要進(jìn)一步研究。
多種重要的藥物都可作為CYP2C8的底物,包括抗癌藥物、抗心律不齊藥物、降血糖藥物、抗癲癇藥物、HMG-CoA還原酶抑制藥物(他汀類藥物)等,紫杉醇是CYP2C8的特征性底物[39]。紫杉醇(paclitaxel)是臨床常用的抗腫瘤藥,主要用于卵巢癌、乳腺癌及非小細(xì)胞肺癌的治療,紫杉醇轉(zhuǎn)化為其主要代謝產(chǎn)物6-羥基紫杉醇是由CYP2C8主要負(fù)責(zé)[40]。AEE對CYP2C8的活性有一定的誘導(dǎo)作用。結(jié)果表明,AEE與紫杉醇聯(lián)用,可能會提高紫杉醇的抗腫瘤效果。
目前,關(guān)于藥物之間相互作用的研究,主要集中在藥物對肝臟藥物代謝酶的誘導(dǎo)或抑制階段,很少有關(guān)于被誘導(dǎo)或被抑制的肝臟藥物代謝酶在停藥后的自我恢復(fù)性研究。基于此,本試驗還考察了AEE對被抑制或被誘導(dǎo)的肝臟藥物代謝酶停止給藥后的影響。根據(jù)SD大鼠和C57小鼠的結(jié)果可以得出:停藥2周后,發(fā)現(xiàn)只有CYP2C19的活性沒有恢復(fù),而其余藥酶活性都有一定的恢復(fù),與空白對照組無顯著性差異(P>0.05)。CYP2C19酶的活性不僅僅與遺傳多態(tài)性相關(guān),還與其他一些外在因素相關(guān)。目前為止,已經(jīng)發(fā)現(xiàn)CYP2C19基因中存在30多個等位基因的突變體[40]。其中CYP2C19*1是編碼正常酶活性的野生型。CYP2C19*2、CYP2C19*3為主要突變基因型,所表達(dá)的酶活性減低[42]。 AEE對CYP2C19的活性具有抑制作用,且該酶的活性在停藥后2周仍未恢復(fù)。原因可能是服用AEE后引起CYP2C19基因發(fā)生了突變,從而導(dǎo)致CYP2C19酶活性降低,其具體的作用機(jī)制有待進(jìn)一步研究。因此,AEE的臨床使用,可能導(dǎo)致動物肝臟藥物代謝酶的抑制或誘導(dǎo),但停止用藥后短期內(nèi)可以恢復(fù)。結(jié)果表明,可以通過停止服用AEE,從而避免其與其他藥物間的相互作用。
綜上所述,本研究結(jié)果表明,AEE在臨床使用中應(yīng)注意由CYP450酶介導(dǎo)而可能發(fā)生的藥物間的相互作用。臨床使用AEE與其他藥物合用時,要特別注意可能出現(xiàn)的藥效學(xué)改變或出現(xiàn)的不良反應(yīng)。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學(xué)大眾(2021年6期)2021-07-20 07:42:44
科學(xué)(2020年3期)2020-11-26 08:18:30
學(xué)苑創(chuàng)造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學(xué)生學(xué)習(xí)指導(dǎo)(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學(xué)學(xué)報(自然科學(xué)版)(2015年2期)2016-01-10 08:41:55
云南中醫(yī)學(xué)院學(xué)報(2014年3期)2014-07-31 18:57:34
