黃精酒制前后多糖單糖組成及抗氧化活性對比研究?
2023-03-07 13:42:12魯文潔陳井太宋志前萬曉瑩彭詩濤劉振麗寧張弛
中國中醫基礎醫學雜志 2023年2期
魯文潔,陳井太,宋志前,萬曉瑩,彭詩濤,劉振麗,寧張弛△,王 淳△
(1.中國中醫科學院中醫基礎理論研究所,北京 100700;2.北京四方中藥飲片有限公司,北京 100072;3.上海林清軒生物科技有限公司,上海 201600)
蒸法是中藥傳統炮制中常用的關鍵技術之一,能增加一些中藥的免疫活性,如黃精、何首烏、五味子、牛膝等[1-4],而多糖類成分是這些中藥表現免疫調節作用的共性有效成分之一[5,6]。由于多糖的結構極其復雜,解析其結構就要先分析其單糖組成[7]。高效液相色譜(High Performance Liquid Chromatography,HPLC)法聯合不同檢測器是目前最常用的單糖組成分析方法。根據所用檢測器的不同,可分為衍生化后測定和直接測定兩種情況。一是聯用二級管陣列檢測器(Diode Array Detector,DAD)或熒光檢測器(Fluorescence Detector,FLD)時,需對多糖水解液進行衍生化后方能測定。1-苯基3-甲基-5吡唑啉酮(3-Methyl-1-phenyl-2-pyrazolin-5-one,PMP)是常用的單糖衍生化試劑。二是聯用蒸發光散射檢測器(Evaporative Light Scattering Detector,ELSD)或示差折光檢測器(Refractive Index Detector,RID)等時,可不進行衍生化處理直接測定單糖[8]。
本文從黃精多糖的單糖組成入手,通過對比HPLC-ELSD、HPLC-RID和PMP-HPLC-DAD 3種分析方法的檢測效果,確定黃精多糖單糖組成的測定方法。并進一步考察黃精酒制前后多糖的單糖組成變化,為明確酒制對黃精多糖初級結構的影響提供依據。同時還對酒制前后黃精多糖的抗氧化活性進行測定,為臨床用藥提供依據。
1 材料
1.1 藥物與試劑
果糖(貨號:100231-201807,純度≥99.6%)、巖藻糖(貨號:112014-201902,≥99.6%)、半乳糖(貨號:100226-201807,≥95.9%)、D-無水葡萄糖(貨號:110833-200904,100%)和鼠李糖(貨號:11683-201502,100%)對照品購自中國食品藥品檢定研究院;D-甘露糖(貨號:C16J8H28561,純度≥98%)、L(+)-阿拉伯糖(貨號:20179462701,純度≥98%)對照品購自上海源葉生物科技有限公司;三氟乙酸(貨號:20190527)購自天津市大茂化學試劑廠;磷酸氫二鉀(貨號:030517)購自北京化學試劑公司;氫氧化鈉(貨號:20120109)購自北京化工廠;1-苯基3-甲基-5吡唑啉酮(3-Methyl-1-phenyl-2-pyrazolin-5-one,PMP,貨號:C11370802)購自上海麥克林生化科技有限公司;1,1-二苯基-2-三硝基苯肼(1,1-Diphenyl-2-Picrylhydrazyl,DPPH,貨號:STBB0719)購自美國sigma公司;乙腈、甲醇為色譜純,其他試藥為分析純。
從多花黃精主產地湖南、安徽、貴州、湖北、江西、廣東等地購買了11個批次藥材,經北京中醫藥大學中藥學院劉春生教授分別鑒定為百合科植物多花黃精Polygonatum cyrtonema Hua,樣品信息見表1。

表1 黃精藥材品種和來源信息
1.2 主要儀器
1200型高效液相色譜儀(含G-1322A在線真空脫氣機,G-1311A高壓四元泵,G-1313A標準自動進樣器,G-1316A恒溫箱,G1315CDAD SL檢測器,G-1362A示差折光檢測器,Agilent HP化學工作站),美國Agilent公司;2000型蒸發光散射檢測器,美國Alltech公司;XWK-Ⅲ型空氣發生器,天津市津分分析儀器制造有限公司;UGC-24C型氮吹儀,北京優晟聯合科技有限公司。
2 方法與結果
2.1 黃精和酒黃精飲片的制備
按照課題組前期確定的工藝條件分別制備得到黃精和酒黃精飲片[9]。取黃精藥材2 kg,洗凈,切厚片(2~4 mm),置于鼓風干燥箱中,45 ℃干燥至含水量15%以下,得到黃精飲片。取黃精飲片1 kg,加黃酒200 g,拌勻,悶潤過夜至酒吸盡;置蒸鍋內隔水加熱,燉至16 h,取出,同上干燥處理,得到酒黃精飲片。
2.2 黃精和酒黃精多糖的制備
取黃精和酒黃精飲片約500 g,加10倍量水回流提取2次,每次1 h,濾過,減壓濃縮(60 ℃),加水定容至500 mL,緩慢加入乙醇,待含醇量達到90%,4 ℃冷藏過夜,離心,棄上清,用90%乙醇洗滌沉淀3次,冷凍干燥,稱重,分別得黃精、酒黃精的粗多糖100.97 g、70.12 g,計算得率分別為20.15%和14.02%。分別取上述黃精和酒黃精的粗多糖,加水制成約1 g·L-1粗多糖溶液,按粗多糖溶液-Sevage試劑(3∶1)加入Sevage試劑[三氯甲烷-正丁醇(4∶1)],劇烈振搖,離心,棄去中間的蛋白層和下層有機層,重復3次,收集上層水液,加乙醇至醇濃度為90%,4 ℃冷藏過夜,離心棄去上清液,沉淀用90%乙醇洗滌3次后,冷凍干燥,得到純化后多糖,稱重,得黃精多糖和酒黃精多糖重量分別為74.21 g、51.76 g,計算得率分別為14.84%和10.35%[9]。
2.3 對照品溶液的制備
取果糖、葡萄糖、阿拉伯糖、巖藻糖、甘露糖、半乳糖和鼠李糖對照品適量,精密稱定,分別加蒸餾水制成每1 mL含11.04 mg、5.60 mg、5.19 mg、5.24 mg、5.49 mg、5.38 mg、5.06 mg的單糖儲備液。精密吸取各單糖儲備液適量,置于10 mL容量瓶中,加蒸餾水稀釋至刻度,即得每1 mL含果糖3.31 mg,葡萄糖0.56 mg,阿拉伯糖0.52 mg,巖藻糖0.52 mg,甘露糖0.55 mg,半乳糖0.54 mg,鼠李糖0.51 mg的混合對照品溶液。
2.4 黃精多糖酸水解條件的優化
2.4.1 因素水平表的設計 以水解溫度、水解時間、酸濃度作為考察因素,采用L9(34)正交表進行三氟乙酸(Trifluoroacetic Acid,TFA)水解黃精多糖的正交試驗設計。以單糖含量為指標,確定黃精多糖的最佳酸水解條件。因素水平見表2。

表2 黃精多糖酸水解正交試驗因素水平表
2.4.2 供試品溶液的制備 取編號為PC-6的黃精多糖約10 mg,9份,精密稱定,置于10 mL西林瓶中,按正交試驗因素表加入不同濃度的TFA溶液2 mL,封蓋,在不同溫度下反應,水解液用氮氣吹干,用2 mL甲醇溶解,氮氣吹干,反復3次,直至TFA完全除盡。沉淀加水溶解,并定容至5 mL容量瓶中,即得。每組平行3次。
2.4.3 色譜條件 色譜柱:Agilent Carbohydrate NH2柱(4.6 mm×250 mm,5 μm)。柱溫:30 ℃;流動相:乙腈-水(75:25);流速:1 mL/min;ELSD漂移管溫度:110 ℃;氣體流量:2.5 L/min;增益:8。
2.4.4 測定法 精密吸取混合對照品和樣品溶液各5 μL,進樣,計算單糖含量。
黃精多糖水解條件優化結果見表3,為避免單糖含量高低影響正交試驗結果,采用權重法對4個單糖含量進行歸一化處理,以綜合評分M為評價指標,對黃精多糖水解條件進行優化,具體公式如下:
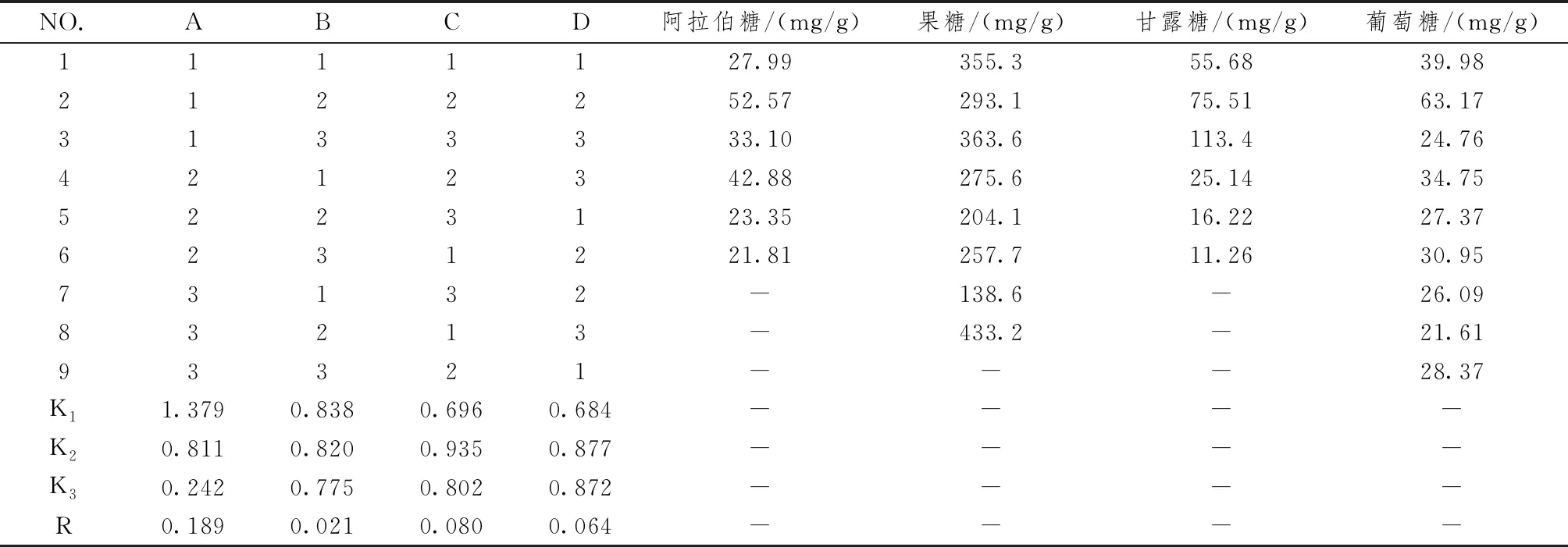
表3 黃精多糖水解正交試驗結果
式中A、B、C、D分別為不同酸水解條件阿拉伯糖、果糖、甘露糖和葡萄糖含量。
由表4可以看出,影響黃精多糖酸水解效果的各因素水平作用分別為A>C>B,即水解溫度>酸濃度>水解時間,但僅因素A(水解溫度)具有顯著性意義(P<0.05)。對于各指標,因素A和因素B均以第1水平最佳,因素C以第2水平最佳,結合方差分析和正交試驗結果,最終確定黃精多糖的最佳水解條件為A1B1C2,即選取1 mol/L三氟乙酸在60 ℃下水解1 h。

表4 方差分析表
2.5 不同單糖檢測方法的對比
對比HPLC-ELSD、HPLC-RID和PMP-HPLC-DAD 3種單糖分析方法的檢測效果,確定最佳的單糖含量測定方法。
2.5.1 供試品溶液的制備 分別取編號為PC-6的黃精和酒黃精多糖約10 mg,精密稱定,置于10 mL西林瓶中,按最佳水解條件進行水解,水解液采用氮氣吹干,加2 mL甲醇溶解,氮氣吹干,反復3次,直至TFA完全除盡。沉淀加水溶解,定容至5 mL容量瓶中,即得。
2.5.2 HPLC-ELSD法分析單糖組成
2.5.2.1 色譜條件 同2.4.3項。
2.5.2.2 測定法 精密吸取對照品和供試品溶液各5 μL,進樣,測定。結果見圖1,黃精和酒黃精多糖中均可檢測到阿拉伯糖、果糖、甘露糖和葡萄糖4個單糖成分。

1.鼠李糖;2.巖藻糖;3.阿拉伯糖;4.果糖;5.甘露糖;6.葡萄糖;7.半乳糖
2.5.3 HPLC-RID法分析單糖組成
2.5.3.1 色譜條件 RID檢測器溫度:40 ℃。其余同2.4.3。
2.5.3.2 測定法 精密吸取對照品和供試品溶液各20 μL,注入液相色譜儀,測定。結果見圖2,黃精和酒黃精多糖均僅檢測到果糖。
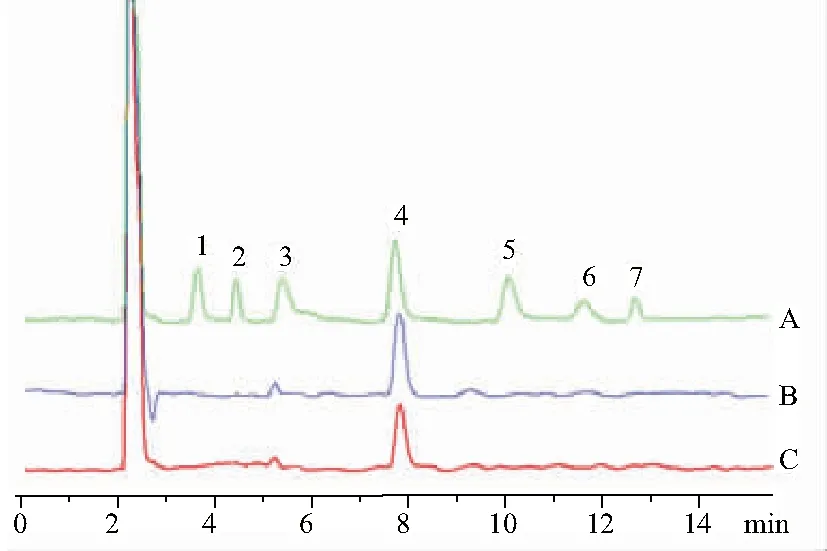
1.鼠李糖;2.巖藻糖;3.阿拉伯糖;4.果糖;5.甘露糖;6.葡萄糖;7.半乳糖
2.5.4 PMP-HPLC-DAD法分析單糖組成
2.5.4.1 色譜條件 色譜柱:Waterssymmetry C18色譜柱(3.0 mm×150 mm,3.5 μm)。柱溫:30 ℃;流動相:乙腈-0.05 mol/L磷酸鹽緩沖液(pH 6.92)(13:87);流速:0.5 mL/min;DAD檢測波長:250 nm。
2.5.4.2 衍生化反應 精密吸取2.3項下混合對照品溶液和2.5.1項下供試品溶液各200 μL,置于試管中,依次加入0.3 mol/L的NaOH溶液和0.5 mol/L的PMP甲醇溶液各500 μL,混合,置于80 ℃下反應1.5 h,冷卻至室溫,再加入0.3 mol/L的HCl溶液500 μL中和反應液,用2 mL三氯甲烷萃取3次,除去PMP,棄去有機層,水層加水定容至2 mL容量瓶中,即得。
2.5.4.3 測定法 精密吸取衍生化后的對照品和供試品溶液各20 μL,進樣,測定。結果見圖3,黃精和酒黃精多糖中均只檢測到葡萄糖和阿拉伯糖。

1.鼠李糖;2.半乳糖醛酸;3.葡萄糖;4.半乳糖;5.阿拉伯糖;6.巖藻糖
對比3種分析方法,HPLC-ELSD法可檢測到阿拉伯糖、果糖、甘露糖和葡萄糖4種單糖;PMP-HPLC-DAD法僅檢測到葡萄糖和阿拉伯糖2種單糖;HPLC-RID法僅能檢測到果糖。因此,HPLC-ELSD法的檢測效果最好,后續實驗采用HPLC-ELSD進行含量測定。
2.6 黃精和酒黃精多糖單糖成分的含量測定
采用HPLC-ELSD法建立單糖的含量測定方法,并對比酒制前后黃精中4種單糖含量的變化。
2.6.1 對照品溶液的制備 分別精密吸取阿拉伯糖、果糖、甘露糖、葡萄糖的對照品儲備液,置于10 mL容量瓶中,加水稀釋至刻度,制成每1 mL含阿拉伯糖、果糖、甘露糖和葡萄糖質量濃度為0.52 mg、3.31 mg、0.55 mg和0.56 mg的混合對照品溶液。
2.6.2 供試品溶液的制備 取各批次黃精和酒黃精多糖約10 mg,精密稱定,置于10 mL西林瓶中,按2.5.1項下方法制備供試品溶液,即得。
2.6.3 方法學考察
2.6.3.1 線性關系的考察 分別精密吸取2.2項下的混合對照品儲備液,按1、2、4、6、8、10倍比進行稀釋,得系列混合對照品溶液,按2.6.1項下的色譜條件進行測定,以峰面積的自然對數(natural logarithm)為縱坐標(Y),相應的質量濃度的自然對數值log C為橫坐標(X),繪制標準曲線,計算回歸方程與線性范圍,求r值。結果如表5所示,各單糖成分相關系數r均達到0.999 2以上,表明4個單糖成分的線性關系良好。

表5 線性關系考察結果
2.6.3.2 精密度試驗 精密吸取編號為PC-6黃精多糖供試品溶液5 μL,連續進樣6次,按2.6.1項下色譜條件測定。結果表明4個單糖成分的峰面積對數值(log A)的RSD均<1.0%,表明該方法的精密度良好。
2.6.3.3 穩定性試驗 精密吸取編號為PC-6黃精多糖供試品溶液5 μL,分別于制備后0、2、4、6、8、24 h測定,結果表明4個單糖成分峰面積對數值(log A)的RSD均<1.0%,表明4個單糖成分在24 h內穩定性良好。
2.6.3.4 重復性試驗 取藥材編號為PC-6的黃精多糖約10 mg,6份,精密稱定,按2.6.2項下方法制備,分別測定并計算4個單糖成分的含量。結果顯示,4個單糖成分含量的RSD均<2.0%,表明該方法重現性良好。
2.6.3.5 回收率試驗 取已知各單糖含量的PC-6黃精多糖約5 mg,6份,精密稱定,分別精密加入果糖、阿拉伯糖、甘露糖和葡萄糖對照品溶液,按2.6.2項下方法制備,分別測定并計算回收率。結果如表6所示,阿拉伯糖的回收率范圍為95.83%~102.9%,RSD為2.63%;果糖的回收率范圍為95.17%~102.6%,RSD為2.45%;甘露糖的回收率范圍為96.36%~104.6%,RSD為3.37%;葡萄糖的收率范圍為97.02%~99.70%,RSD為1.10%。4個單糖回收率值的RSD均<5.0%,表明該方法回收率符合標準。

表6 加樣回收率考察結果
2.6.4 測定法 精密吸取黃精和酒黃精多糖供試品溶液5 μL,注入液相色譜儀,測定,即得。結果如表7所示,11批黃精多糖的單糖含量范圍分別為阿拉伯糖39.59~97.48 mg/g,果糖249.2~570.2 mg/g,甘露糖40.39~98.19 mg/g,葡萄糖54.82~95.43 mg/g;11批酒黃精多糖的單糖含量范圍分別為阿拉伯糖0.00~37.18 mg/g,果糖127.6~348.8 mg/g,甘露糖0.00~61.0 mg/g,葡萄糖65.76~106.4 mg/g;果糖在黃精酒制前后均為主要的單糖成分。同黃精相比,同批次酒黃精的果糖、甘露糖、阿拉伯糖的含量均降低,葡萄糖含量有所升高。其中5批酒黃精多糖未檢測到阿拉伯糖,7批黃精多糖未檢測到甘露糖。

表7 黃精多糖的單糖含量結果(mg/g)
2.6.5 主成分分析 采用Simca-P14.1軟件對黃精和酒黃精的單糖含量進行主成分分析法(Principal Components Analysis,PCA)處理,共提取出3個主成分,其累積解釋率R2(cum)為0.967,預測能力Q2(cum)為0.783,其中,其中,第一主成分解釋了54.9%的變量信息,第二主成分解釋了26.8%的變量信息,第三主成分解釋了15.0%的變量信息。如圖4所示,黃精和酒黃精可明顯區分,表明炮制對黃精的單糖含量影響較大。
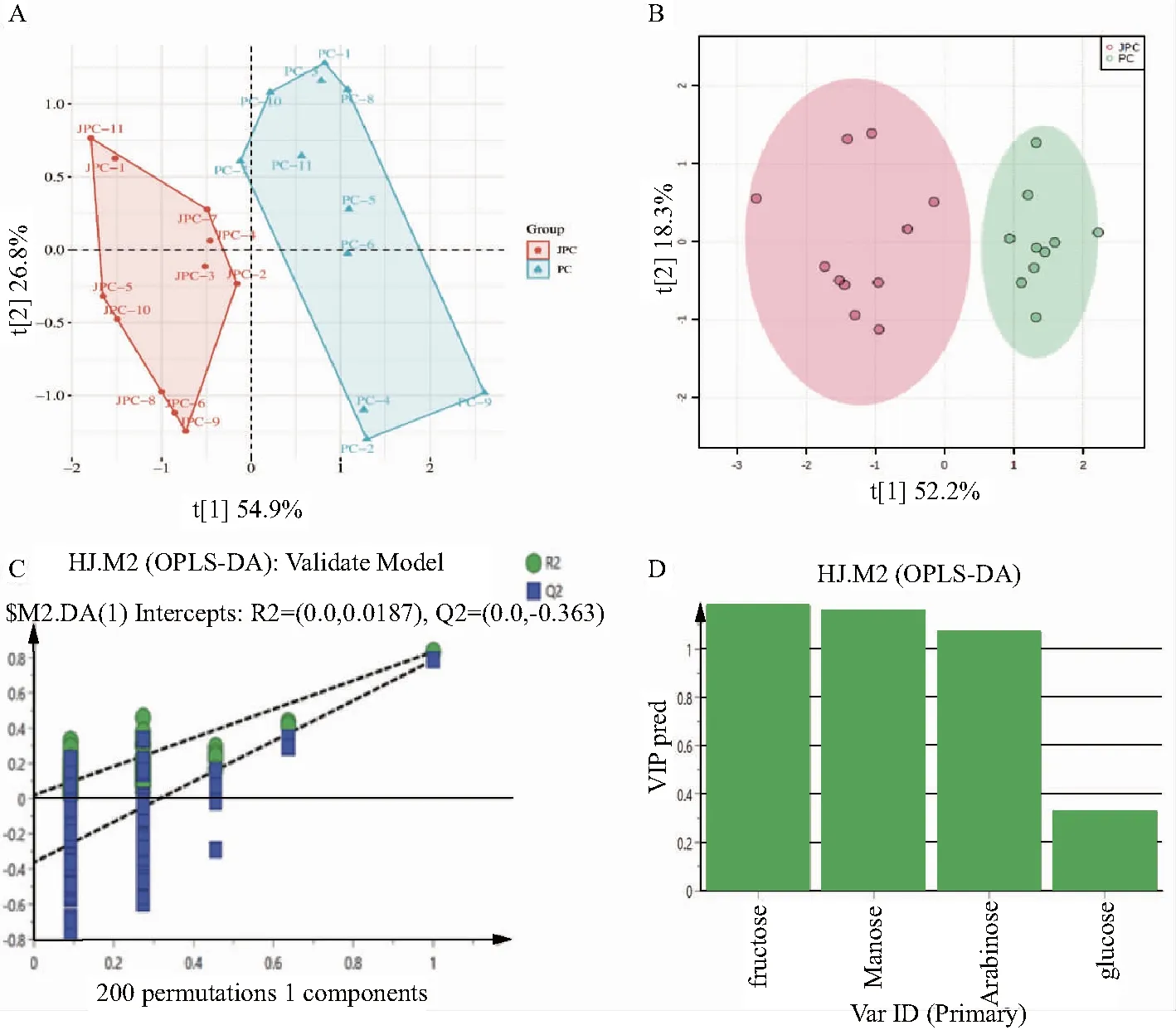
注:A.PCA得分圖;B.OPLS-DA得分圖;C.置換檢驗圖;D.VIP值圖
2.6.6 正交偏最小二乘法判別分析 為進一步考察黃精和酒黃精的差異,在PCA無監督分析的基礎上,開展有監督的正交偏最小二乘辨別分析法(Orthogonal Partial Least Squares-Discriminant Analysis,OPLS-DA)分析,并對其進行置換檢驗,以驗證該模型的擬合程度,結果見圖4。OPLS-DA得分圖顯示,其R2X為0.914,R2為0.833,Q2為0.786,提示模型具有較高的可信度以及模型預測能力強。進一步對OPLS-DA模型參數R2和Q2進行了200次的置換驗證,結果顯示R2Y的截距未超過0.3,Q2Y的截距未超過0.05,表明OPLS-DA模型可靠,可用于下一步的分析。以變量重要性投影(VariableImportance inProjection,VIP)>1為標準篩選差異化合物,結果顯示黃精與酒黃精的差異化合物為阿拉伯糖、甘露糖和果糖,經酒制后三者含量均降低。
2.7 黃精酒制前后多糖的抗氧化活性變化
參考文獻的方法,用無水乙醇配制0.1 mmol·L-1DPPH溶液,避光保存[10,11]。分別將不同濃度提取物及2 mL DPPH溶液加入同一試管中,搖勻,室溫下靜置30 min后測定其吸光度A2,同時測定DPPH溶液與2 mL溶劑混合后的吸光度A0,以及提取物與2 mL溶劑混合后的吸光度A1。按照{抑制率=[1-(A2-A0)/A1]×100﹪}計算清除率,清除率越大抗氧化能力越強。
分別取不同濃度的黃精和酒黃精粗多糖,測定DPPH自由基清除率,計算半抑制濃度IC50,結果見圖5。
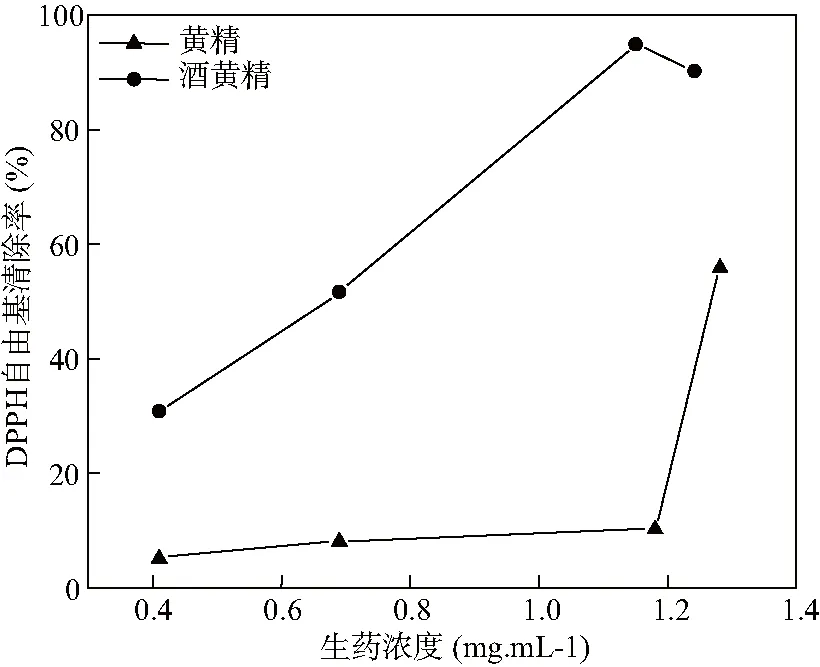
圖5 不同濃度多花黃精和酒黃精多糖的DPPH自由基清除率(n=3)
3 討論
單糖組成分析是研究中藥多糖組成、結構、作用及構效關系的基礎[12]。目前常用的檢測方法是多糖酸水解成單糖,再分析其單糖組成。有研究表明單糖水解反應的速率與其種類相關,且過高的水解溫度會產生副產物影響測定結果[13]。因此,優化酸水解條件對于分析多糖的單糖組成是非常重要的。此外,不同的檢測器對多糖單糖組成的影響也較大,目前常用的有HPLC-ELSD法、HPLC-RID法和PMP-HPLC-DAD法[14]。PMP-HPLC-DAD法常用于檢測酸性、中性和堿性單糖,但需對單糖進行衍生化處理[15]。HPLC-RID法和HPLC-ELSD法可直接用于測定單糖,無須衍生化,但重復性較差[16]。本研究對3種方法進行比較,結果顯示,HPLC-ELSD法可測到阿拉伯糖、果糖、甘露糖和葡萄糖;PMP-HPLC-DAD法僅檢測到葡萄糖和阿拉伯糖,推測其未測到果糖原因是PMP僅能與醛糖發生衍生反應,未檢測到為酮糖的果糖[17];甘露糖的保留時間過短,可能與PMP的色譜峰重疊。HPLC-RID檢測法的靈敏度較差,故僅測到含量較高的果糖。因此,HPLC-ELSD法最適合用于檢測黃精和酒黃精多糖的單糖組成。此外,本文未檢測到文獻報道的鼠李糖、半乳糖醛酸、乳糖和巖藻糖[18,19],推測其原因可能與品種有關。
炮制后中藥的多糖含量會顯著降低,而單糖含量上升[20]。本文對比了黃精和酒黃精的單糖組成和含量,結果顯示,酒制前后均是以果糖為主的雜多糖,但酒制后果糖、阿拉伯糖和甘露糖含量下降,且部分酒黃精多糖未檢測到阿拉伯糖和甘露糖,葡萄糖含量有所上升。推測在高溫高濕的炮制環境中,多糖發生水解,且部分單糖可能發生麥拉德反應,使飲片發生褐變[21],如阿拉伯糖和果糖。此外,DPPH自由基清除結果顯示,黃精炮制后IC50值降低,抗氧化活性增強,提示上述黃精酒制前后抗氧化活性的增強可能與單糖組成的變化有關。
