SHP2及其變構抑制劑在腫瘤靶向治療中的研究進展
2023-03-01 01:33:48李媛李雪陳真
藥學研究 2023年1期
李媛,李雪,陳真
(1.中國藥科大學藥學院,江蘇 南京 211198;2.南京圣和藥業股份有限公司,江蘇 南京 210038)
細胞進行各項生命活動依賴于復雜的信號網絡,信號蛋白的磷酸化和去磷酸化是常見的翻譯后活性調節方式,分別由蛋白激酶和蛋白磷酸酶調控。真核生物中磷酸化位點常發生在絲氨酸、蘇氨酸和酪氨酸上,其中蛋白酪氨酸激酶(protein tyrosine kinase,PTK)和蛋白酪氨酸磷酸酶(protein tyrosine phosphatase,PTP)共同調節胞內蛋白酪氨酸磷酸化水平,調控蛋白功能發生轉變,使信號向下傳遞[1]。
根據國際癌癥研究機構發布的全球癌癥2020年統計報告(GLOBOCAN 2020)推算可知[2],新發癌癥約為1 930萬例,癌癥死亡人數近1 000萬例。傳統癌癥治療手段不僅給患者帶來極大的痛苦和不良反應,而且無法達到比較滿意的治療效果。隨著腫瘤分子生物學的發展,科學家們認識到可用藥物靶向腫瘤細胞上基因異常或過表達的致癌分子,在不影響正常組織細胞的生命活動情況下,可實現優于細胞毒性藥物的抗腫瘤效果,且不良反應更小。目前已有多種激酶抑制劑被用于癌癥治療,例如第三代EGFR抑制劑奧希替尼、CDK4/6抑制劑帕博西尼、mTOR抑制劑依維莫司、BCR-ABL抑制劑伊馬替尼、ALK抑制劑艾樂替尼等。而靶向致癌蛋白磷酸酶則提供了另一種治療思路。含Src同源2結構域蛋白酪氨酸磷酸酶2(SHP2)是PTP家族中第一個[3]報道的致癌蛋白,參與多種癌癥的發生發展過程,經過20年的開發,目前已由催化位點抑制劑轉向變構位點抑制劑,發揮“分子膠水”的作用,穩定SHP2的閉合構象。
1 SHP2蛋白結構與功能
SHP2是一種由PTPN11基因編碼的胞內非受體蛋白酪氨酸磷酸酶,其結構包含兩個N端SH2結構域(N-SH2和C-SH2),以及C端具有催化功能的PTP結構域和一個富含脯氨酸基序并帶有酪氨酸磷酸化位點(Tyr542和Tyr580)的C端尾巴[4]。X射線晶體衍射結果表明[5],在基礎狀態下,SHP2通過N-SH2與PTP結構域相互作用阻礙其催化活性,使該磷酸酶處于自抑制構象;當存在細胞因子、生長因子等刺激時,酪氨酸磷酸化蛋白與SH2結構域結合,暴露PTP結構域的催化活性位點,解除磷酸酶的自抑制狀態,激活SHP2。
SHP2介導多條信號通路,包括RTK/MAPK、PI3K/AKT、JAK/STAT、PD-L1/PD-1等[4,6-7],參與機體正常發育[8]、心血管生成[9]和免疫應答[10],調節細胞生長、分化、增殖、存活和凋亡等生理活動。SHP2還可通過調節谷氨酸受體的表達或功能參與突觸可塑性、學習和記憶過程,其功能喪失型突變(loss of function,LOF)會導致豹綜合征(LEOPARD syndrome,LS),而獲得型突變(gain of function,GOF)會引發努南綜合征(Noonan syndrome,NS),兩綜合征具有發育遲緩和學習困難的重疊癥狀[11-12]。除此之外,SHP2的異常還與腫瘤密切相關,其突變在腫瘤中發生頻率較低,但常常因基因過度表達[13-15]或被異常激活使信號通路增強,促進腫瘤的發生發展[16]。
2 SHP2在腫瘤靶向治療中的價值
2.1 SHP2在多種腫瘤中發揮促癌作用 已有研究證實[17],PTPN11是白血病、肺癌和乳腺癌的癌基因,為了研究該基因在黑色素瘤中的作用,Hill等[17]利用PTEN和CDKN2A缺失驅動的小鼠黑色素瘤模型發現PTPN11在黑色素瘤中被激活,并通過促進非錨定集落形成和腫瘤生長產生促癌作用。另有研究顯示[18]SHP2在宮頸癌中表達上調,一方面,在Hela和SiHa細胞中敲除SHP2,細胞生長和遷移受到抑制,對順鉑的敏感性增加;另一方面,SHP2過表達與淋巴結轉移和高HPV DNA有關,促進宮頸癌的發生發展。Yang等[19]在膠質母細胞瘤細胞中導入突變體SHP2 E76K,該激活突變體在體外增強了細胞增殖、遷移和侵襲能力,在異種移植模型上產生促癌作用。上述研究均可證明SHP2自身的過度激活(如表達上調或功能獲得型突變)可促進癌癥的發生發展。
受體酪氨酸激酶(receptor tyrosine kinase,RTK)過度激活將直接導致下游多條信號通路過強,驅動癌癥發生發展,而 SHP2是連接RTK和下游多條信號通路的關鍵節點,是致癌途徑的必經之處,抑制SHP2可產生明顯的抗腫瘤效果[20-22]。PDGFRα信號激活的膠質母細胞瘤由于化療耐藥和血腦屏障,治療極具挑戰性,SHP2變構抑制劑SHP099能抑制PDGFR下游效應因子JUN,減弱細胞周期進程,與神經祖細胞相比,SHP099在體外優先抑制膠質瘤干細胞樣細胞的存活和自我更新,在體內展示了顯著的生存獲益[20]。Zhao等[21]對ErbB2轉基因小鼠條件性敲除SHP2,并抑制HER2擴增乳腺癌細胞株的SHP2,在兩種乳腺癌模型上的研究結果均表明,SHP2是ErbB2誘導腫瘤發生、腫瘤組織發育所必需的,條件性敲除SHP2可阻斷ErbB2過表達、誘導正常細胞表型、抑制腫瘤發生和轉移。表達受體酪氨酸激酶FLT3-ITD突變并且帶有DNA甲基化調節因子TET2或DNMT3A功能缺失突變的急性髓細胞白血病小鼠模型概括了人類白血病的基本特征,SHP099可抑制白血病細胞生長并誘導其分化,下調癌基因Myc信號,使白血病細胞中與細胞增殖和自我更新相關的基因表達程序正常化[22]。
2.2 SHP2參與腫瘤耐藥 骨髓基質細胞(bone marrow stromal cells,BMSCs)的保護作用使B細胞急性淋巴細胞白血病(B-ALL)對化療耐藥,研究發現,PTPN11在初診和復發的B-ALL的BMSCs中高表達,使用質粒誘導BMSCs中SHP2激活,顯著增加了其介導的CCRF-SB細胞對長春新堿的耐藥,實驗結果顯示SHP2通過PI3K/AKT通路上調血管細胞黏附分子-1(VCAM-1),其與黏附分子VLA-4的相互作用誘導黏附效應,進而造成化療耐藥[23]。
大多數接受TKI治療的非小細胞肺癌(non-small cell lung cancer,NSCLC)患者常發生耐藥,最終經歷疾病進展,獲得型耐藥是臨床主要挑戰,其機制依賴于EGFR突變或是不依賴EGFR的旁路激活,而無論哪一耐藥機制最后仍需通過中介信號分子SHP2向下傳遞至RAS/MAPK通路。最近報道的SHP2抑制劑IACS-13909(13)已證實其在奧希替尼耐藥模型的治療價值,包括通過CRISPR-cas9技術引入EGFR C797S突變的EGFR L858R/T790M/C797S NCI-H1975細胞,該模型對奧希替尼敏感度下降,以及含EGFR ex19del 的HCC4006-奧希替尼耐藥細胞模型,該模型經驗證未產生耐藥突變,研究顯示聯用可以延緩耐藥的發生,增加對奧希替尼的敏感度[24]。
在RAS/MAPK信號通路依賴腫瘤中,用RAF和MEK抑制劑阻斷該信號往往引起RTK的重新激活和ERK信號的反彈,引起腫瘤對藥物的適應性抵抗,而SHP2介導了這一負反饋[25-26]。RAF常發生V600E突變,突變后激酶活性增加500倍,導致下游MEK-ERK 信號通路持續激活。單獨使用其特異性抑制劑維羅非尼往往出現耐藥性,Putlyaeva等[25]以高表達BRAF V600E的甲狀腺濾泡上皮為模型,研究維羅非尼治療后應用siRNA介導PTPN11表達下調,結果顯示,一方面參與腫瘤耐藥性形成的CCNA1和NOTCH4基因表達下調,另一方面,參與細胞周期調控的基因如p21、p15、p16、rb1和IGFBP7的轉錄活性下調。MEK位于ERK上游,其抑制劑曲美替尼因可負反饋上調p-SHP2,導致快速產生耐藥性因而作為單藥的應用受到限制,而SHP2抑制劑SHP099阻止耐藥產生的作用在多種癌癥包括KRAS突變和野生型RAS的腫瘤中得到證實,因此與MEK抑制劑聯用可成為防止對激酶抑制劑耐藥的廣泛的治療策略[26]。
2.3 SHP2參與免疫抑制信號 SHP2在多種免疫細胞中均有表達,主要參與抑制性受體(inhibitory receptors,IRs)下游信號通路,介導免疫抑制[27]。在T細胞中[28],SHP2通過SH2結構域與程序性細胞死亡-1(PD-1)二聚體上的兩個pITSM-Y248殘基相互作用,誘導酶的激活,進而促進T細胞受體(TCR)信號復合物中ZAP70激酶在PD-1胞漿尾SHP2募集后的去磷酸化作用,同時介導CD28信號失活,從而使T細胞增殖、分化受到抑制,干擾素(IFN)產生減少,抗腫瘤免疫功能下降。在巨噬細胞中[27],SHP2在巨噬細胞集落刺激因子-1(CSF-1)刺激下與其誘導的信號蛋白復合物Grb2/Gab1結合,激活RAS-ERK通路,促進巨噬細胞增殖和M2型極化。當巨噬細胞表面的銜接蛋白SIRPα與其配體CD47結合后,為SHP2的去磷酸化招募特異性底物,以抑制細胞內信號傳導和減少巨噬細胞的吞噬作用,幫助腫瘤逃避宿主免疫監視。NK細胞[27]表面的IRs也可以通過ITIM基序招募和激活SHP2,產生免疫抑制作用。
為驗證抑制SHP2在體內能產生抗腫瘤免疫作用,研究人員用對SHP099不敏感的CT-26結腸癌細胞分別在裸鼠和免疫系統完整的小鼠體內建立異種移植模型,實驗結果顯示SHP099對裸鼠的腫瘤生長影響最小,而在免疫完整小鼠體內CD8+IFN-γ+T細胞增加,顆粒酶B和穿孔素等細胞毒性T細胞相關基因表達上調,降低腫瘤負荷,且在SHP2條件性敲除T細胞的小鼠體內腫瘤生長明顯減慢,證實了SHP099與PD-1阻斷可發揮相輔相成的作用[29]。80%的晚期NSCLC患者常因內在或獲得性抵抗對單獨給予免疫檢查點抑制劑不響應,然而,在SHP2抑制劑和PD-L1抗體聯合XRT放射線治療抗PD-1耐藥的344SQ NSCLC的129Sv/Ev小鼠研究中,顯示出良好的系統性抗腫瘤效應,增強局部和遠處反應,減少肺轉移,提高小鼠生存率,這是因為XRT增加了遠處腫瘤SHP2+M1 TAMs,SHP099的使用與高M1/M2比例、CD8+T細胞增加、調節性T細胞降低有關[30]。此外,目前利用巨噬細胞調節腫瘤微環境免疫主要存在兩大困難,一是癌細胞分泌的巨噬細胞集落刺激因子(MCSF)與巨噬細胞上的受體CSF-1R結合,可誘導腫瘤相關巨噬細胞(TAMs)向免疫抑制的M2表型分化;二是癌細胞過表達的跨膜蛋白CD47與髓系細胞上的信號調節蛋白SIRPα連接后,激活胞內SHP1和SHP2,進而激活“eat-me-not”信號通路,吞噬功能受到抑制。Ramesh等[31]設計并合成了一種裝載CSF-1R和SHP2抑制劑的納米粒靶向M2型巨噬細胞,體外結果顯示M1復極化和吞噬功能增強,在體內高侵襲性4T1乳腺癌模型和B16F10黑色素瘤模型上,與單藥相比次優劑量給藥效果更佳且沒有毒性,提示SHP2抑制劑與靶向免疫系統藥物聯用是一種有前景的治療策略。
總的來說,SHP2的異常活化在大多數腫瘤中具有促癌作用,作為RTK的下游信號分子傳導多條致癌通路,參與耐藥和免疫逃逸的發生,是治療惡性腫瘤有前景的靶點,并且也是當前的研發熱點。十余年的研究旨在發現可成藥的SHP2抑制劑,抑制腫瘤進展、延緩耐藥出現、增強腫瘤免疫應答。
3 SHP2變構抑制劑的開發進展
近20年的研究確定了SHP2在胚胎發育和致癌中的作用,但沒有同時具有有效性、成藥性、特異性的SHP2抑制劑走上臨床。2016年之前的第一代抑制劑占據PTP催化口袋,阻止底物去磷酸化,盡管活性最優可低至0.2 μmol·L-1,但由于催化位點帶正電荷的極性環境,抑制劑帶負電荷才能有足夠的親和力結合并抑制其活性,這勢必導致化合物細胞透膜性差、口服生物利用度低[32]。且PTP之間催化域的高度同源性,特別是與SHP1,這導致第一代抑制劑選擇性差,無法特異性靶向SHP2[33]。隨著2016年諾華發現SHP2的“隧道狀”變構位點,抑制變構磷酸酶SHP2轉向了一個全新的方向,且獲得審批進入臨床試驗階段的SHP2變構抑制劑越來越多。目前已經發現3個變構位點可穩定SHP2的閉合構象,隧道樣位點位于C-SH2、N-SH2和PTP結構域的表面,門閂樣和溝槽樣位點分別位于N-SH2和PTP結構域表面的兩側[34]。本文重點介紹了近5年文獻中報道的SHP2變構抑制劑。
3.1 諾華SHP2變構抑制劑及其衍生物 2016年諾華[33]首次報道了通過高通量篩選和X射線晶體學識別到的突破性化合物——作用于SHP2變構結合口袋的SHP836,經結構優化后確定SHP099(1)是一種可同時滿足有效性、特異性、生物利用度良好的SHP2抑制劑(酶抑制活性IC50=0.07 μmol·L-1),其與隧道樣變構位點結合,穩定SHP2自抑制構象進而使催化位點處于持續封閉狀態。該化合物的細胞增殖抑制活性、溶解度、選擇性、藥代動力學性質及體內藥效等性質良好,使其成為一個較優的工具化合物來研究SHP2在生理病理中的作用,亦可進一步優化產生更優化合物。2018年該研究團隊[35]利用可干擾SHP099結合的工程雙突變SHP2 T253M/Q257L細胞株進行了新的篩選,發現了第二種變構抑制劑SHP244(2),該變構結合位點不同于SHP099的隧道狀位點,而是位于N-SH2和PTP表面的裂隙。SHP244(2)是SHP2弱抑制劑,酶抑制活性IC50=60 μmol·L-1,水溶性差(在pH 6.8緩沖液中為0.047 mmol·L-1),親脂性高(clog P=3.9)。基于結構設計優化后,酶抑制活性、水溶性及熱穩定性等均有提高,但其活性仍低于SHP099。X射線晶體結構驗證了該變構位點抑制劑并未干擾SHP099的結合,因此聯合應用兩個變構位點抑制劑能夠增強細胞的通路抑制,證明雙變構抑制是可能的。
2019年[35]進一步發現了多個5,6融合雙環支架,占據同樣的隧道樣變構位點,經過結構變形和優化后識別到吡唑嘧啶類代表化合物SHP389(3)及14(4)衍生物。其抑制活性均在亞微摩爾級別,同時對30個GPCRs、離子通道、核受體、轉運體、酶和激酶的IC50>30 μmol·L-1。SHP389(3)對hERG有微弱的抑制作用,詳細的Q-Patch表明其不參與功能性hERG相互作用。但由于滲透性和暴露量差,不能進一步評價其體內藥效。14(4)雖具有明顯的心臟毒性(IC50=0.29 μmol·L-1),但表現出良好的PKPD相關性。緊接著,該研究團隊[36]發現了結構更多樣的SHP2變構抑制劑,將上述融合雙環變形為新穎的單環嘧啶酮支架,發現6-氨基3-甲基嘧啶類化合物SHP394(5)為強效、選擇性和口服有效的SHP2抑制劑,具有良好的物理化學和ADME特性。但滲透性差,Caco-2細胞通透性[Papp(A-B)1.02×10-6cm·s-1]較低,明顯外排(B-A/A-B=15),這可能解釋了其藥代動力學研究中中度口服暴露量和較低生物利用度(F=29%),因此需進一步優化提高其藥代動力學性質。
TNO155(6)是諾華[37]也是世界上第一個走上臨床的SHP2變構抑制劑,目前與EGFR-TKI藥物、CDK4/6抑制劑、KRAS G12C抑制劑、免疫療法聯合使用評估其臨床效果。TNO155(6)抑制酶(IC50=0.011 μmol·L-1)、p-ERK細胞信號通路(IC50=0.011 μmol·L-1)及細胞增殖(IC50=0.1 μmol·L-1)的活性達到有史以來最低,BCS I類性質使其生物利用度達到100%,小鼠異種移植瘤模型中10 mg·kg-1即具有明顯抑瘤效果。在評估的4個臨床前物種中,均能觀察到中低清除率、較早到達Tmax(0.8~2 h)、中度血漿蛋白結合率(61%~81%),與體外微粒體和肝細胞觀察到的低清除率及物理化學性質相符。低濃度時TN0155(6)不抑制CYP3A4、2D6或2C9,因此聯合使用其他藥物時發生藥物-藥物相互作用的風險最小。
在諾華報道的有效化合物基礎上,研究人員展開了更進一步的研究。基于SHP836發現的compound 1(7)[38]抑制SHP2活性的IC50值為9.97 μmol·L-1,對BaF3細胞的IC50值為10.73 μmol·L-1。熒光滴定實驗證實其與SHP2直接結合,分子動力學模擬研究顯示其對SHP2的親和力明顯高于SHP1,與SHP836相比,不僅具有相似的抑制作用,而且與SHP2結合后能形成更加穩定的構象。癌癥相關的SHP2突變大多位于N-SH2:PTP結構域界面[39],E76、A72、D61/G60和E69是N-SH2結構域中突變最頻繁的氨基酸殘基。為探究SHP099 是否抑制SHP2突變體,研究人員在用TF-1人白血病細胞構建的4種常見突變(p.D61Y、p.E69K、p.A72V和p.E76K)細胞中,SHP099能有效抑制TF-1 SHP2 E69K細胞的生長,IC50為(1.46±0.46)μmol·L-1,降低了p-ERK1/2和抗凋亡蛋白BCL-XL,并誘導DNA修復酶PARP裂解。在SHP099(1)的基礎上[40],采用支架跳變的方法設計新型SHP2抑制劑,11a(8)是吡啶類系列衍生物中最有效、最具選擇性的SHP2抑制劑,其直接與SHP2蛋白結合,體外酶活IC50為1.36 μmol·L-1,對SHP1的活性超過100 μmol·L-1,抑制Ba/F3細胞IC50為2.35 μmol·L-1。ADMET預測分析證實其具有良好的類藥性質。具體見圖1。
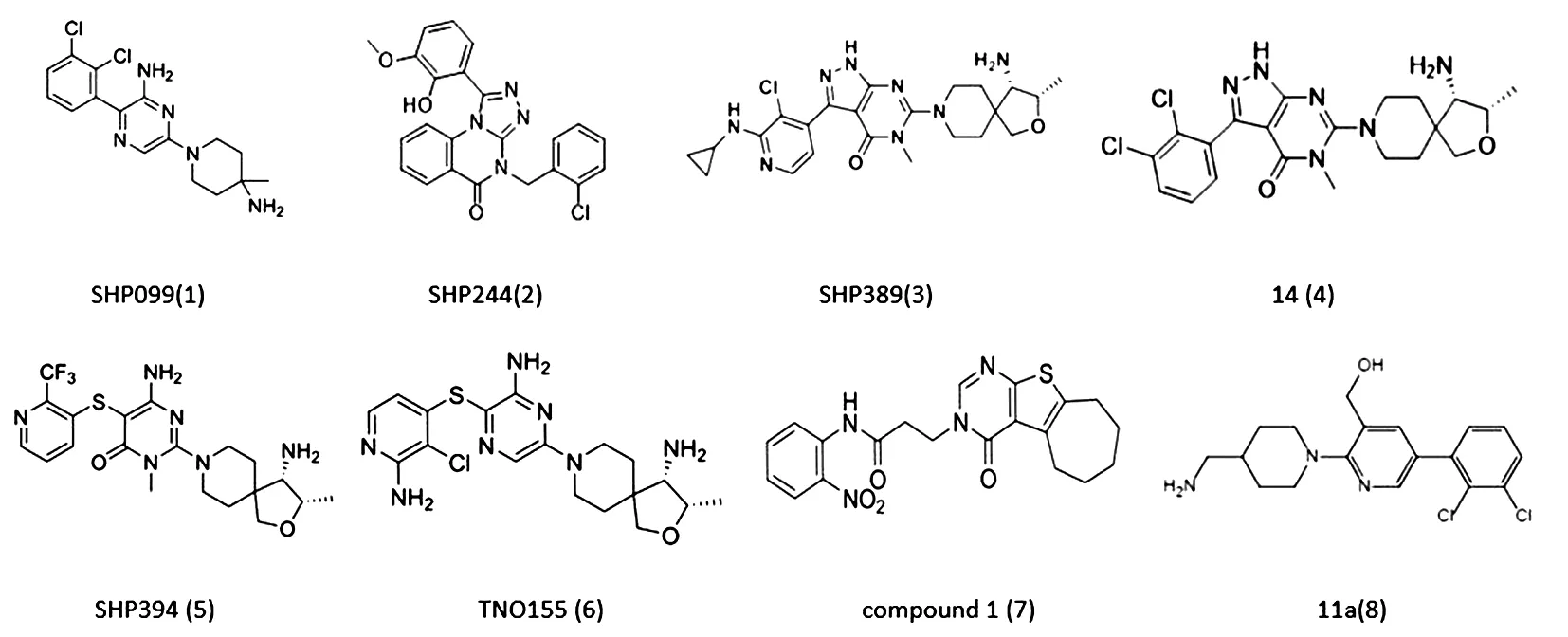
圖1 諾華SHP2變構抑制劑及其衍生物
3.2 其他SHP2變構抑制劑 SHP2 E76位于N-SH2:PTP橋接處,突變之后自抑制構象不穩定,用GOF SHP2 E76A建立的新的篩選策略鑒定了含2-氨基噻唑支架的化合物23(9),與隧道樣變構位點結合,對SHP2 E76A的IC50為0.7 μmol·L-1,對其他PTP具有至少30倍選擇性優勢,而(1)對SHP2 E76A的IC50更低(0.12 μmol·L-1)。23(9)不僅抑制GOF SHP2 N58S NCI-H661細胞、EGFR T790M H1975細胞、FLT3-ITD突變的白血病細胞系如MV4;11和MOLM-13增殖,還能拮抗YAP轉錄活性,提示YAP活性受SHP2 PTP催化功能調節。23(9)在小鼠上具有可接受的藥代動力學特性和體內藥效[41]。
研究人員利用高通量篩選(HTS)技術發現了苯并噻唑嘧啶支架,可同時結合C-SH2和PTP結構域來變構抑制致癌磷酸酶。優化后得到的類似物2(10)是一種具有選擇性的競爭性變構抑制劑,該化合物既不干擾SHP2 PTP結構域活性,也不干擾SHP2的F285S、T253M、Q257L突變,該變異體包含兩個在變構位點上的突變,使得在空間上阻止化合物進入SHP2。但是對急性髓系白血病細胞MOLM-14的活性很差(IC50=85±8 μmol·L-1)[42]。多肽化合物具有易于修飾和快速合成,無毒性,且比重組抗體免疫原性低的特點。盡管存在溶解性差、膜通透性差等缺點,但仍有明顯的優勢,如作用效高、靶選擇性好、在組織中的積累少等。例如二肽化合物Phe-Asp(11)可在微摩級別(IC50=5.2±0.4 μmol·L-1)抑制SHP2,對PTP1B有10倍的選擇性,5 μmol·L-1顯著降低MCF7細胞活力[43]。
位于SHP2 C-SH2和PTP表面的位點與同源SHP1相比表現出特定的特征,假設與該位點結合的化合物可能會加強C-SH2和PTP結構域的連接,穩定SHP2封閉的自抑制構象,以此通過計算機輔助藥物設計(CADD)篩選虛擬化合物數據庫,在10個候選化合物中LY6(12)活性最優,SHP2活性抑制IC50為9.8 μmol·L-1,而對SHP1的IC50為72 μmol·L-1。LY6(12)對PTPN11條件敲除(PTPN11fl/fl/Ade-Cre+)的小鼠胚胎成纖維細胞(MEFs)不敏感,證實了SHP2是該化合物的主要靶標。攜帶SHP2激活突變的腫瘤細胞比WT細胞對LY6(12)更敏感(例如SHP2 E76K突變體IC50值為7.67 μmol·L-1)。SHP2激活突變相關的青少年粒單細胞白血病(JMML)對粒細胞-巨噬細胞集落刺激因子(GM-CSF)和IL-3表現出超敏反應,Ba/F3細胞被IL-3刺激后,ERK、AKT、JAK2和STAT5的活化被LY6(12)抑制[44]。
IACS-13909(13)是SHP2直接的變構抑制劑,酶活性抑制IC50=15.7 nmol·L-1,具有高度選擇性,對PTP結構域IC50大于50 μmol·L-1。X射線晶體結構顯示SHP2變構結合袋的Pro491與IACS-13909(13)的吡唑吡嗪環相連,該位點突變會阻止化合物結合,SHP2 P491Q過表達則顯著降低了IACS-13909(13)的敏感性。IACS-13909(13)能有效抑制RTK基因改變及對TKI或RTK sh RNA敏感的細胞株,但對BRAF V600突變細胞系及大多數KRAS突變細胞系不敏感,不能抑制其p-ERK或p-MEK。目前IACS-13909(13)的衍生物IACS-15509正在臨床上進行評估[24]。
PCC0208023(14)可以非競爭性地抑制全長SHP2酶的活性(IC50=2.10 nmol·L-1),其活性優于RMC-4550兩倍,但對SHP2的自由催化結構域缺乏活性,這與變構模式的抑制一致。在4種KRAS驅動的結直腸癌細胞(LS180、HCT116、SW1463、SW837)中,PCC0208023(14)的抑制作用同樣優于RMC-4550。其在LS180腫瘤給藥24 h后仍保持高水平,組織上主要分布于腸道和肺部,且與RMC-4550暴露量相似,在HCT116模型30 mg·kg-1劑量下,二者均能顯著抑制腫瘤重量、推遲腫瘤生長,免疫組化顯示Ki67和p-ERK水平降低,腫瘤中cleaved caspase-3表達增加,凋亡增加,但是都產生了明顯的體重下降[45]。
為了鑒定新的具有潛在抗癌活性的SHP2抑制劑,研究人員以DiFMUP為底物篩選了658種天然產物,重樓皂苷D(15)是從傳統藥用植物重樓中分離,可選擇性地變構抑制SHP2,酶抑制活性為15.3 μmol·L-1。據報道,重樓皂苷D(15)可誘導線粒體跨膜電位去極化,導致H2O2生成、細胞色素C釋放和凋亡誘導因子的產生。與外周血單個核細胞(PBMC)相比,SHP2在人白血病中過表達,特別在Jurkat細胞中表達水平最高,細胞抑制活性為2.8 μmol·L-1,降低胞內p-ERK水平,增加裂解PARP水平,導致細胞凋亡死亡[46]。具體見圖2。
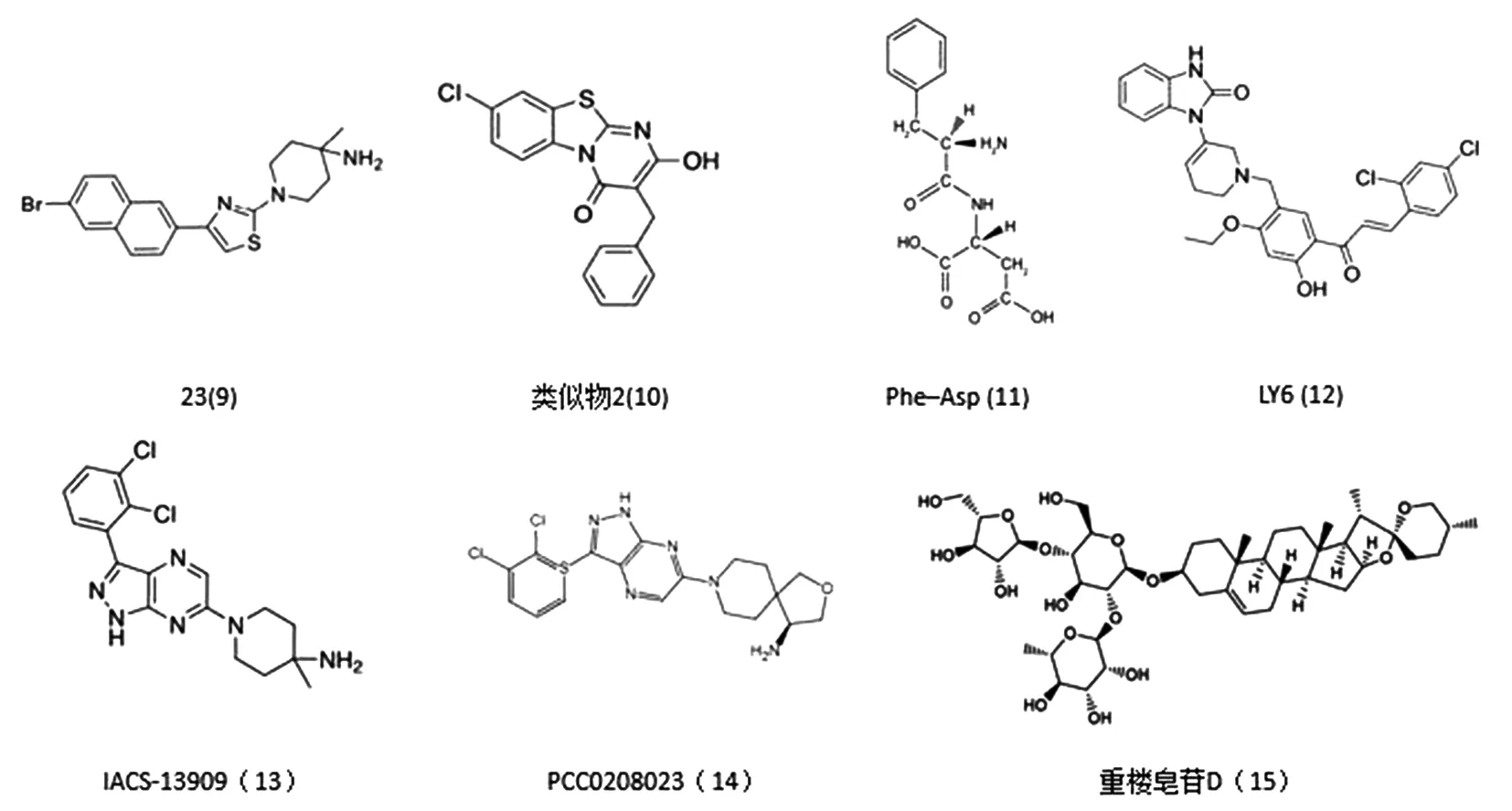
圖2 其他SHP2變構抑制劑
諾華的TNO-155,加科思的JAB-3068、JAB-3312,Revolution Medicines的RMC-4630以及Hoffmann-La Roche的RLY-1971屬于早期走進臨床試驗的SHP2變構抑制劑[47],目前均處于1/2期招募狀態,以單獨用藥或聯合EGFR突變抑制劑奧希替尼、納扎替尼,MEK1抑制劑考比替尼,KRAS G12C抑制劑JDQ443、阿達格拉西布,PD-1抗體Spartalizumab,CDK4/6抑制劑瑞博西尼,及ERK1/2抑制劑LY3214996,在非小細胞肺癌、結直腸癌、乳腺癌等復發、難治、轉移性實體瘤中評價安全性。近兩年,Navire Pharma的BBP-398(IACS-15509)、Erasca的ERAS-601、南京圣和藥業的SH3809、奕拓醫藥的ET0038及輝瑞的PF-07284892也相繼被開發進入臨床試驗。同時多個候選化合物目前處于臨床前研究階段,包括BT-102、SNG-201、ICP-189、HBI-2376。
4 總結與展望
盡管SHP2變構抑制劑的發現克服了催化位點抑制劑的缺點,但變構位點上的突變(例如D61、E76、A72)卻產生了類似耐藥的結果[48]。近年來發展起來的特異性敲除相關蛋白的化學工具—蛋白水解靶向嵌合體(proteolysis-targeting chimeras,PROTACs)可能是應對SHP2變構位點突變體的一種可選擇方式。一般來說,PROTACs是雙功能分子,包括與靶蛋白結合的配體和與E3泛素連接酶結合的配體,這兩個配體通過一個連接子共價連接,從而召集E3連接酶誘導目標蛋白的泛素化和隨后的降解[49]。基于此原理設計的化合物SP4在納摩爾級別有效抑制Hela細胞生長,其活性是SHP099的100倍,降解胞內SHP2蛋白水平,誘導細胞發生凋亡,展示了誘導SHP2蛋白降解的優勢[49]。另外,針對SHP2的廣泛分布和底物的多樣性,考察SHP2抑制劑的副作用和毒性尤為重要,利用靶向藥物遞送制劑例如納米制劑來規避其對正常組織的影響,可能是一種行之有效的方式。
本文概述了近些年SHP2在腫瘤中的研究進展,總結了其抑制劑特別是變構抑制劑的發展概況,本文將有助于藥物開發學者認識該靶點現況及技術攻關難題,研發具有市場前景的藥物,未來理想的靶向腫瘤細胞、腫瘤浸潤淋巴細胞及腫瘤血管[50]的SHP2變構抑制劑將會聯合多種科學技術實現精準治療。
猜你喜歡
保健醫苑(2022年5期)2022-06-10 07:46:38
現代臨床醫學(2022年3期)2022-06-06 07:59:40
昆明醫科大學學報(2022年1期)2022-02-28 07:43:40
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
科學大眾(2020年12期)2020-08-13 03:22:22
中國生殖健康(2019年3期)2019-02-01 06:12:26
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
中國當代醫藥(2015年17期)2015-03-01 02:03:58
