大學生研究性教學實踐
——以芳香烴核磁共振的理論計算為例
2023-01-10 05:27:30張春芳劉子揚趙紫琳凡綠潔張攢商艷麗霍樹營馬剛
大學化學 2022年12期
張春芳,劉子揚,趙紫琳,凡綠潔,張攢,商艷麗,霍樹營,馬剛
河北大學化學與環境科學學院,化學國家級實驗教學示范中心,河北 保定 071002
為提高學生的創新精神和解決問題的能力,我們以學生“學”為中心,在課堂實踐、畢業設計和第二課堂中不斷嘗試問題導向教學、項目式教學和研究性教學等多種教學模式改革的探索[1-3]。在不額外增加課時的前提下,著重延展和探索了研究型和創新型教學內容的應用,從而增加了課程的挑戰度、創新性、高階性。其中,研究性教學以學生為主體,結合教師科學研究中的內容、技術和方法,引導學生學習和實踐新內容和新方法,達到了知識學習、思維鍛煉和能力提高等綜合目標[4],也促進了學生解決實際問題以及創新能力的培養。
核磁共振(nuclear magnetic resonance,NMR)是研究有機物結構的重要手段。然而,作為大型儀器,所提供的上機時間有限,從而限制了學生對NMR的深入認識和解析。隨著計算機等技術的發展,基于文獻參數的數據驅動法(經驗)和理論計算模擬等方法研究NMR日趨完善[5]。其中理論計算與模擬,因其便捷和高精度的特點在研究分子結構和性質中具有重要的意義。本文中,我們以芳香烴NMR的理論計算為例,介紹了第二課堂中的研究性教學過程。具體流程如表1所示,主要包括:針對教學內容和目標對學生進行分組;學生分工進行文獻調研、概念總結和技術探索等活動;教師指導學生對示例物質進行建模和理論計算;學生對目標物質建模計算并分析數據;學生總結結果并書寫報告;教師評價并反思教學。上述研究性教學的實施旨在促進學生掌握NMR的基本原理和技術、提高譜圖解析能力,也有助于激發學生學習興趣、提高其自主學習能力。

表1 研究性教學內容和流程
1 文獻調研及內容總結
1.1 NMR發展應用
NMR是非零磁矩的原子核在外磁場作用下自旋能級分裂的一種現象,可提供豐富的物質結構和動力學信息。自NMR被報道以來,其分析檢測理論和儀器設備日趨完善,在化學、物理、生物、地質、醫療以及材料等領域的教學科研[6,7]中發揮了巨大作用。近年來,高場磁體、連續相調制多脈沖和動態核極化等新技術不斷引入固體NMR,使得人們對凝膠、液晶、晶體以及非晶態等不同物質結構的認識更加深刻[8]。
理論計算方法和技術在微觀物質結構構建和性質預測方面具有獨特的優勢,其中密度泛函理論預測的NMR化學位移具有足夠的準確性,在物質結構解析和性質預測方面具有一定的價值。中國科學院化學研究所已經構建了基于密度泛函理論的有機分子化學位移在線計算平臺[9],為物質結構的解析和認識提供了支持。
1.2 化學位移
NMR反映了自旋量子數非零的原子核在外磁場作用下的能級躍遷。不同的物質結構的核外電子排布不同,其產生的感生磁場對原子核的屏蔽效應也不同,進而導致相同原子核在外磁場中吸收頻率的改變。相對于同一參比物的磁吸收頻率(核磁屏蔽張量)的變化被稱為化學位移。化學位移受多種因素影響,包括電子誘導和共軛作用、碳原子雜化、磁各向異性、氫鍵和范德華作用等[10]。此外,溫度、溶劑等因素也會影響化學位移。目前,在教學和科研中,通常是通過液體NMR技術測定13C和1H化學位移,并據此判斷化學環境、推測物質結構信息。相比于液體NMR,固體NMR能夠判定聚集形態、相等獨特信息[11]。然而,固體狀態中分子運動受限,偶極-偶極相互作用(dpole-dipole interaction)、化學位移各向異性(chemical shift anisotropies)以及四極矩耦合(quadrupolar coupling)等導致化學位移譜線嚴重展寬,分辨率下降[12]。雖然實際操作中有多種方法可以消除上述相互作用,對于復雜的物質結構,化學位移譜峰的歸屬仍十分困難、繁瑣。目前,教學科研中仍主要依靠經驗解析化學位移進而判斷物質結構。從特定結構的模型構建和化學位移模擬等理論與計算模擬角度出發,有助于經濟、有效地解析物質結構并分析物質性質。
1.3 自旋-自旋耦合常數
除化學位移之外,高分辨率NMR譜線分裂的裂距,即自旋-自旋耦合常數(通常以J表示),也能夠反映電子結構和空間結構的獨特信息。J反映了原子核耦合作用的強弱,通常,兩個原子核相隔n個化合鍵數目的耦合常數表示為nJ,如nJ(H,H)、nJ(C,H)和nJ(C,C)。相比于化學位移,J受外磁場影響較小,也可以判定結構,特別是構象結構[13]。如Klepach等[10]使用3J(C,H)和1J(C,C)研究了雙鍵和三鍵的立體化學;Tuttle等[14]通過H原子間的自旋-自旋耦合常數明確了生物大分子的氫鍵;Cremer等[15]通過C原子間自旋-自旋耦合常數,定量評估了C-C鍵;Ambati和Rankin[16]通過有機基團中Si、H自旋-自旋耦合常數研究了硅烷中烷氧基的水解程度。因而,理論計算得到J值有助于對分子內和分子間的化學鍵、分子構象和物質結構的立體化學等更精細問題的分析[17]。
2 理論建模及模擬方法總結
2.1 化學位移計算方法
理論計算通過模擬目標物質以及參比物質(通常為四甲基硅烷,TMS)核磁屏蔽張量的差值得到化學位移。常用理論計算方法有原子軌道法(gage-including atomic orbital,GIAO)[18]和連續軌距變換法(continuous set of gage transformations,CSGT)[19]。GIAO法是Ditchfield[18]提出的具有顯式場依賴的基函數的方法。CSGT法是Keith和Bader[19]開發的具有量規不變性、對實空間點進行連續軌距變換控制,從而精確描述屏蔽張量的電流密度的方法。蘇永超等[20]使用Gaussian 03計算了鄰乙酰氧基苯甲酸的化學位移,并比較GIAO和CSGT兩種方法。他們發現用Hatree-Fock方法和密度泛函理論(Density functional theory,DFT)算芳環碳的化學位移時,CSGT法比GIAO法更為準確。為與化學位移的實驗值進一步對比,又有研究人員對理論計算的磁屏蔽常數進行了系數校正處理,即標度法[21]。標度法的表達公式如下:

其中δ為化學位移,a為截距,σ為各向同性磁屏蔽常數值,b為斜率。不同計算水平下,a、b標度參數可以從CHESHIRE CCAT網站選取近似水平的數值[22],σ直接讀取Gaussian 09輸出文件的SCF GIAO Magnetic shielding tensor部分的Isotropic值。
2.2 自旋-自旋耦合常數計算方法
自旋-自旋耦合常數J包含費米接觸(Fermi contact)、自旋偶極(Spin-dipolar)、順磁性自旋軌道耦合(paramagnetic spin-orbit)和反磁性自旋軌道耦合(diamagnetic spin-orbit)四項貢獻。其中費米接觸項為電子與核的相互作用,對J起到最主要貢獻。J值可以通過Gaussian中的NMR = spinspin關鍵詞計算得到,從輸出文件中的Total nuclear spin-spin coupling J (Hz)項中可直接獲取目標原子間的核自旋-自旋耦合常數數值。
2.3 理論計算實踐
本文使用Gaussian 09程序[23]的B3LYP泛函和6-311G**基組對目標分子進行了結構優化和頻率計算,所得結構均有零個虛頻。為明確取代基給受體效應,在B3LYP/6-311G**水平計算了優化結構的自然鍵軌道及二階微擾能(E2);為獲得化學位移,分別在B3LYP/6-311G**和M06-2X/def2-TZVPP水平對優化結構進行NMR計算(分別用b-b和b-m表示)。將NMR計算所得磁屏蔽常數標準物質的磁屏蔽常數對比獲得化學位移;或者將磁屏蔽常數代入公式(1),利用標度法計算化學位移。我們從文獻[22]所列多個表格中選取相近計算水平的數據,獲得兩種標度法的數據:
標度法I (處理b-b的NMR數據,簡稱標I):

標度法II (處理b-m的NMR數據,簡稱標II):
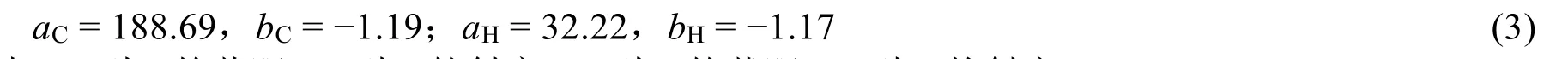
其中,aC為C的截距,bC為C的斜率;aH為H的截距,bH為H的斜率。
3 理論計算實踐及結果分析討論
3.1 氣相芳香烴分子的化學位移討論
我們構建了圖1a所示的芳香族分子的模型,優化了結構并通過二階穩定化能E2和Mulliken電荷分別分析了取代基對苯環整體及每個原子的影響。由圖1b可知,甲酰基、硝基和羧基,皆為吸電子基團,羥基、氨基和乙酰氧基皆為給電子基團。由表2中的電荷數據可知,相同取代基對苯環上不同原子的影響不同。
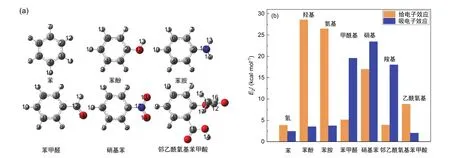
圖1 芳烴分子模型示意圖(a)和吸電子及給電子效應(b)
我們計算了圖1a中芳烴分子的化學位移,匯總于表2,并與SDBS網站[24]展示的數據進行了對比。由表2中數據可知,δC和δH在b-b計算水平與實驗分別相差0.54-11.96和0.02-0.70、在b-m水平相差分別為> 16和0.87-1.44;兩種標度法中,標II與實驗吻合更好。表2中的數據明確展示了吸電子取代基導致與其相連的苯環碳化學位移增大。例如,硝基苯中δ2C(147.23,標II)大于苯中δC(128.77,標II)。對于苯酚、苯胺和苯,氧的電負性大于氮大于氫,電負性強的取代基降低了與取代基相連的碳原子(2C)的核外電子密度(Mulliken電荷分別為0.15、0.10和-0.10),因而苯酚的δ2C(153.29,標II)大于苯胺(148.02,標II)大于苯(128.77,標II);而氧和氮原子上對的孤對電子,對苯環有給電子共軛效應,導致1C和3C化學位移減小。
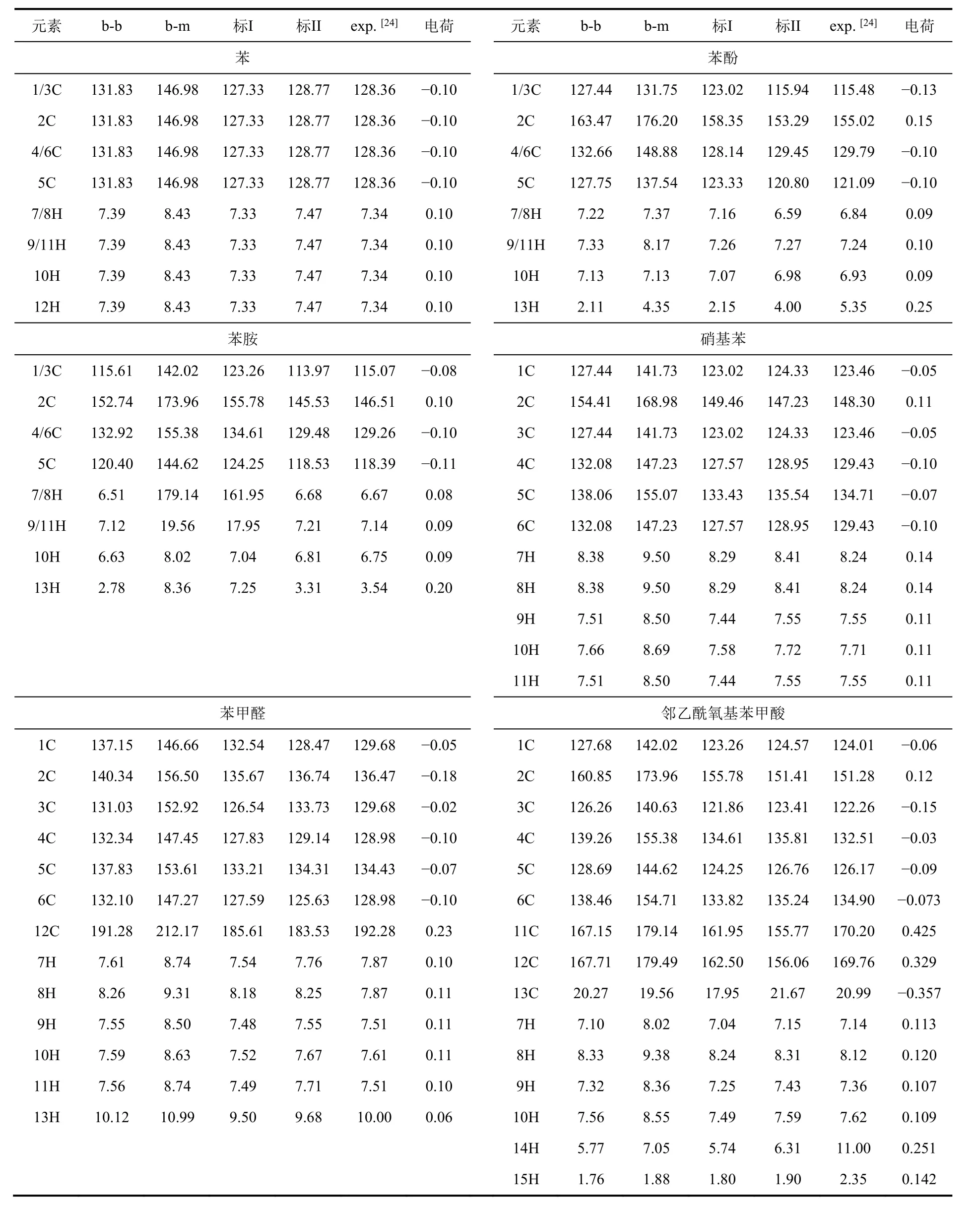
表2 芳香族化合物不同計算和處理方法的化學位移及B3LYP/6-311G**水平的Mulliken電荷
然而,由于理論計算針對單個分子、未考慮分子間氫鍵等復雜作用,其計算數值與實驗值仍有差距,特別是含有羥基的體系,理論計算不能考慮溶液與羥基、羥基之間氫鍵等復雜相互作用。如,苯甲醛中6C和12C的化學位移(125.63和185.81,標II)與實驗值(128.98和192.28)相差較大;鄰乙酰氧基苯甲酸在b-b水平下化學位移的δ12C(167.71)與實驗值(169.76)有一定誤差,標度法校正(161.95)未提高與實驗值的吻合度。類似情況還有,苯酚中羥基氫12H (δH(標I) = 2.15、δH(標II) = 4.00、δH(實驗) =5.35)、鄰乙酰氧基苯甲酸氫14H (δH(標I) = 5.74、δH(標II) = 6.31、δH(實驗) = 11.00)。不過,理論模擬對取代基的影響以及結構定性預測仍有一定的指導意義。
3.2 并五苯不同狀態的化學位移的討論
我們對并五苯分子處于(圖2a)氣態和固態(圖2c)的化學位移進行了計算。氣態計算與3.1節相同,相對于標準物為四甲基硅烷(圖2b)獲得了b-b、b-m、標I和標II等四種數據(如圖3所示)。固態計算使用CASTEP進行,其標準物為金剛烷(圖2d)。
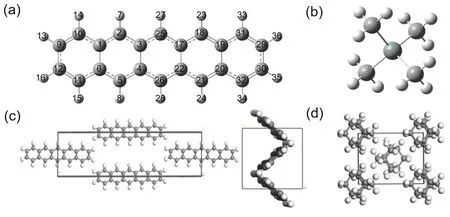
圖2 并五苯分子及原子標號(a)、四甲基硅烷(b)、并五苯晶體正視圖及側視圖(c)、金剛烷固體(d)
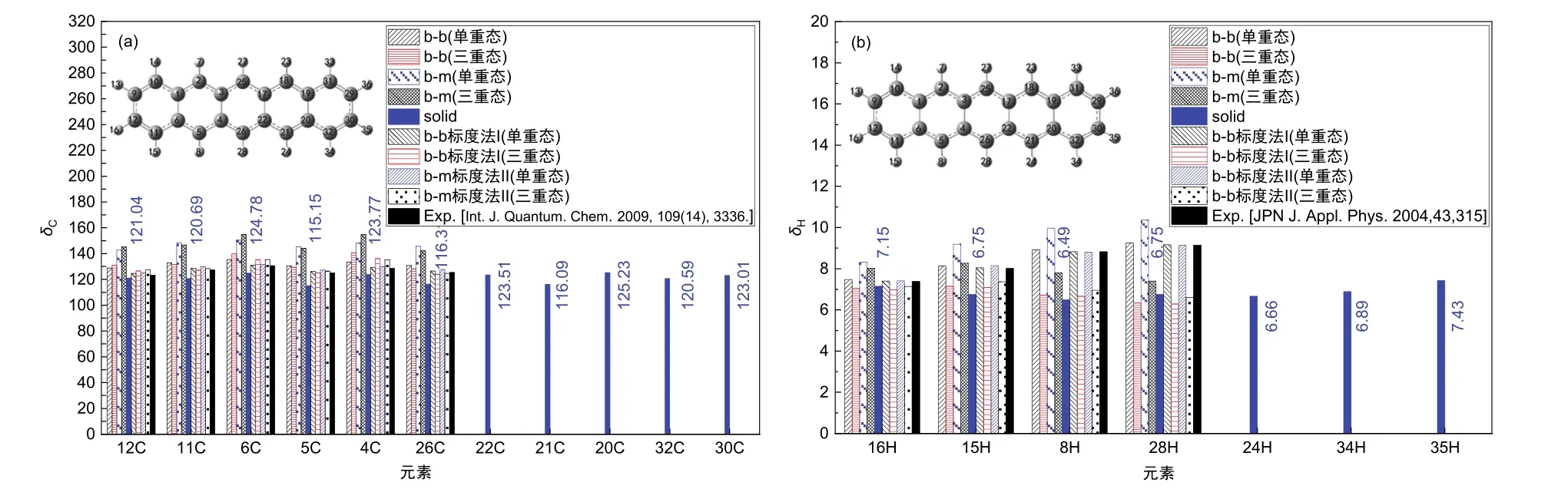
圖3 理論模擬的并五苯體系的化學位移
由圖3可知,C的化學位移經標度法處理后與實驗值[25]更接近,而H的化學位移經標度法處理后與實驗值[26]相差更遠。從圖3還可以看到,單個并五苯分子中對稱分布的C和H的化學位移具有相同的數值,體現了結構對稱性;而對固態并五苯的長軸和短軸C、H原子的化學位移由于周圍環境的差異,數值不相同,特別是長軸上的化學位移數值分布不完全對稱。從圖3還能看到單重態中C的化學位移與三重態相比相差很小,而單重態中H的化學位移始終大于三重態的化學位移,這體現了電子自旋轉變對C原子影響較小、對H原子影響較大。三重態中H電子由自旋相反到自旋平行的轉變使得H原子受到的電子屏蔽效應增加,進而使得三重態中H的化學位移減小。上述化學位移的差異有助于闡明體系的原子結構,也有助于確定其電子態,進而促進結構和性質解析。
3.3 萘與并五苯氣相的自旋-自旋耦合常數討論
我們還計算了萘與并五苯氣相時碳原子間的自旋-自旋耦合常數nJ(C,C),計算結果與萘耦合常數實驗值[27]列于表3。通過比較萘的計算值與實驗值,萘間隔一個鍵的耦合常數計算值間隔一個鍵的自旋-自旋耦合常數1J(C,C)最大(57.63-68.60 Hz)且與實驗值相差較大(6.39-8.20 Hz)。間隔兩個鍵的碳原子,由于單雙鍵間隔以及相互作用的減弱,其自旋-自旋耦合常數2J(C,C)變為負值(-1.85 - -1.19 Hz)。間隔三個鍵的碳原子的自旋-自旋耦合常數3J(C,C)為正值(2.86-8.02 Hz)且3J(C,C) <1J(C,C)。間隔四個鍵的碳原子間相互作用減弱,其耦合常數4J(C,C)為-1.54 - -0.15 Hz。間隔二至四個鍵的耦合常數計算值與實驗值相差較小(0.07-0.60 Hz)。
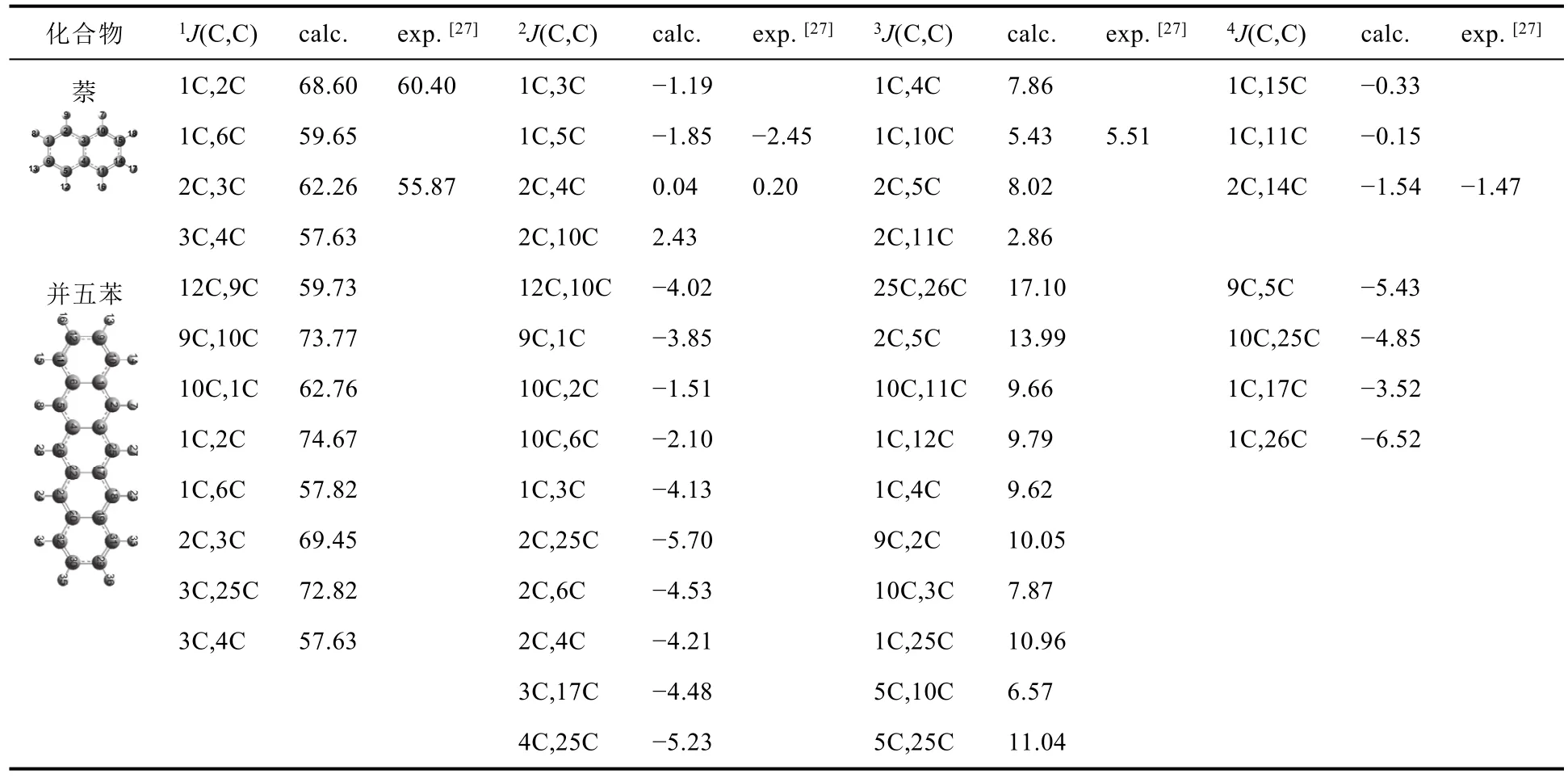
表3 b-b方法所得的并五苯自旋-自旋耦合常數nJ(C,C) (單位:Hz)
對于并五苯,間隔一個鍵的自旋-自旋耦合常數1J(C,C)最大(57.63-74.67 Hz),其中長邊的兩個碳之間的相互作用高于短邊兩個碳之間的相互作用。間隔兩個鍵的碳原子,由于單雙鍵間隔以及相互作用的減弱,其自旋-自旋耦合常數2J(C,C)變為負值(-5.70 - -1.51 Hz),并且邊緣兩個碳原子之間的耦合弱于中心碳原子之間的耦合。間隔三個鍵的碳原子的自旋-自旋耦合常數3J(C,C)受π鍵共軛及多個耦合路徑影響,自旋相互作用變強(6.57-17.10 Hz)[24];最強的為對稱軸上的兩個碳原子(25C、26C)之間的耦合,其次為其平行的碳原子(2C、5C)之間的耦合,再次為與對稱軸兩原子相關(1C、25C或5C、25C)的自旋耦合或同一個六元環內部的碳原子。間隔四個鍵的自旋-自旋耦合常數4J(C,C)為-6.52 --3.52 Hz。
總之,當n為奇數時,萘與并五苯的nJ(C,C)為正值,且隨n的增加,nJ(C,C)的絕對值減小;當n為偶數時,大部分的nJ(C,C)為負值。根據耦合常數的總體趨勢可得出,|1J| > |3J| > |2J| > |4J|,這與文獻[27]和[28]中的結論一致。因而,自旋-自旋耦合常數的正負和大小有助于判斷類似分子的骨架結構,進而準確判定化合物結構。
4 教學成效與反思
在以芳香族化合物的核磁共振模擬計算為例的研究性教學實踐中,學生掌握了目標體系的建模、NMR的計算與模擬的基本方法,完成了化學位移的模擬計算,分析總結了取代基、單重態/三重態、分子結構對化學位移的影響,也探討了原子位置與自旋耦合常數的關聯,從而拓展了對核磁共振原理及其應用的認識。除此以外,學生在研究性學習中做了大量文獻調研和閱讀,提高了有效信息提取和整理的能力;完成了數據總結和報告展示,提高了歸納表達等能力,培養了嚴謹認真和科學探究精神。本項目的實施引起了學生對理論與計算化學的興趣,目前已有兩名學生進入鄭州大學、中國科學院等院校專門從事理論與計算方向深造;累計有十余名學生在創新實驗、大學生創新創業項目等實踐課程中繼續使用所學建模和計算方法。總之,研究性教學活動拓展了學生的知識和技能,提升了學生解決問題和自主學習的能力,研究性教學活動取得了良好的成效。
上述研究性教學過程中,教師積極開展大學生課程教學方法改革,倡導研究性教學理念,重視創新能力培養和科學素質提高。在取得成效的同時,也對此次教學進行了反思,提出了需要改進的方面。例如,上述過程中,部分模擬結果與實驗數據有差別,我們將引導學生以客觀的態度對待這種差別。以實驗數據為準,要求學生進一步研讀文獻,探究實驗條件及設備精度、理論模型不足及計算水平等客觀因素的影響。著重考慮理論計算中存在的諸如忽略氫鍵、范德華相互作用、物體狀態和溶劑效應等因素,以嚴謹客觀的科學態度認識和處理相關狀況。此外,教師在指導學生過程中,應根據不同學生的能力給予不同層次的指導,實現因材施教,發揮特長并拓展能力。教師還應不斷提高組織和管理能力,協調好教師的主導作用和學生的主體地位,管理好不同教學節點和學生活動進展,從而完成研究性教學。總之,教師不僅要緊跟科學研究前沿,還要積極探索教學實踐,不斷推進教學與科研的融合,從而有助于提高學生的綜合素質、有助于為國家培養高素質的創新人才。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
當代陜西(2022年5期)2022-04-19 12:10:18
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:28
湘潮(上半月)(2021年4期)2021-07-20 08:05:28
汕頭大學學報(自然科學版)(2020年4期)2020-12-14 07:05:00
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
科技知識動漫(2017年5期)2017-05-11 21:34:16
科技知識動漫(2017年4期)2017-04-15 22:24:55
科技知識動漫(2017年2期)2017-02-06 20:59:46
科技知識動漫(2016年10期)2016-10-18 20:35:00
