表面結構與催化性能關系的模型催化實驗
——以CuO/Cu2O納米晶催化乙烯實驗與量化計算為例
2023-01-10 05:26:10劉梓歌王炯涵李俍江國順李紅春吳強華張萬群馮紅艷吳紅王鈺熙
大學化學 2022年12期
劉梓歌,王炯涵,李俍,江國順,李紅春,吳強華,張萬群,馮紅艷,吳紅,王鈺熙
化學國家級實驗教學示范中心(中國科學技術大學),合肥 230026
催化劑表面結構與催化性能之間的關系一直是表面催化領域的前沿研究課題。該課題包含了催化反應動力學、熱力學、表面化學、結構化學中重要的物化概念及實驗方法,因此有必要引入到物理化學實驗教學中。而在物化的傳統實驗教學項目中,因為實驗條件的苛刻性或者催化劑表面結構復雜性等原因[1],這一重要課題鮮少被涉及,毫無疑問限制了學生對于催化劑結構及催化過程的進一步理解。近年來,科研工作者提出的基于形貌規整、大小均一、組成可控的納米晶催化劑可以在實際催化反應條件下研究催化劑表面結構和催化性能的方法[2,3],使得將這一前沿課題納入實驗教學成為可能。
通過對催化劑活性進行測試,可獲得催化劑宏觀性能數據;通過量化計算研究催化劑微觀性能,可獲得原子層面上的催化劑表面結構信息和催化反應機理。實驗與量化計算互相驗證和補充,可建立起研究宏觀催化性能與微觀表面結構之間關系的通道。其中,量化計算已經是物理化學研究的必要手段,也是本科物理化學實驗乃至物理化學教學中一個亟待補充完善的內容,我們在本實驗中進行了量化計算與實驗結果對比分析的嘗試。
教學組選取了性質穩定、易于制備[4]的暴露(100)晶面和(111)晶面的立方體和八面體CuO/Cu2O納米晶作為模型催化劑體系,同時基于綠色安全性的考慮,將科研工作中的一氧化碳實驗體系[2,3]改為乙烯作為反應氣的新實驗體系,設計了結合催化劑活性測試和量化計算的創新綜合實驗。實驗中測試了兩種表面結構不同的催化劑對乙烯的催化氧化活性,通過Arrhenius公式計算表觀活化能,判斷兩者表面活性位點的差異。隨后通過搭建CuO/Cu2O晶面結構并判斷活性位點,加強對催化劑微觀結構的認識,由量化計算研究乙烯分子在催化劑表面的吸附情況,結合實驗結果進行分析,進而從微觀上理解表面結構對催化活性的影響。通過對納米晶模型催化劑體系的實驗與量化計算結果的綜合討論,掌握研究催化劑宏觀催化性能-微觀表面結構關系的方式方法。
1 實驗部分
1.1 實驗原理
1.1.1 活性測試實驗
本實驗采用流動法測定固相催化劑的活性,使用轉化率評價催化劑的活性。轉化率指反應物的轉化量占引入反應器反應物總量的百分比。實驗中,反應氣為乙烯與空氣的混合氣體(乙烯/空氣體積比為0.5%)。活性測試實驗試劑、催化劑、實驗儀器詳細信息請見表1-表3。
在200-300 °C的溫度范圍,反應氣能穩定存在。當反應氣經過CuO/Cu2O納米晶催化劑床層時,會發生以下反應:

實驗通過氫火焰離子檢測器(FID)檢測乙烯含量。FID的工作原理是含碳有機物在氫火焰中燃燒時,產生化學電離,在電場作用下,正離子被收集到負極,產生電流,經過放大收集記錄數據。對于在相同的反應條件下(同樣的催化劑裝量,反應物料的進料速度等),根據(1)式計算不同反應溫度下的乙烯轉化率χ。

其中[A]0為反應氣在反應管入口處的乙烯濃度;[A]為出口的乙烯濃度。利用氣相色譜分析,[A]0、[A]分別對應于色譜峰的峰面積S0和S。轉化率又可表示為:

利用Arrhenius公式得到該催化反應的表觀活化能Ea。具體計算過程如下:

即

其中,k為速率常數,Ea為表觀活化能,R為氣體常數,T為熱力學溫度,A為指前因子。

其中,γ為化學反應速率;A’為反應相關的常數。

其中,S為乙烯反應氣體的流量,實驗中在室溫下控制流量為20 mL·min-1,即1200 mL·h-1;η為乙烯體積占比,為0.005;χ為乙烯催化氧化反應的轉化率;ρ為乙烯的密度,標準狀況下的乙烯密度為1.264 g·L-1,利用理想氣體方程計算室溫下(25 °C)的密度為1.158 g·L-1;m為催化劑質量,實驗上稱取0.05 g;Mr為乙烯的相對分子質量。
1.1.2 量化計算
量化計算已成為研究材料性質的重要手段之一,在材料催化機理的理解方面也發揮了越來越重要的作用。使用相關應用程序可實現密度泛函理論數值方法的計算,幫助我們從原子尺度上研究材料表面催化性質。SIESTA (Spanish Initiative for Electronic Simulations with Thousands of Atoms)是一個免費的計算軟件,可用于分子和固體的電子結構計算和分子動力學模擬,基于其內在原理,通常可以在一般的工作站上模擬幾百個原子的體系[5]。
基于自旋極化的密度泛函理論(DFT)方法[6],使用SIESTA軟件包來完成所有的計算。在計算中,采用的是廣義梯度近似方法(GGA)的Perdew-Burke-Ernzerhof (PBE)交換泛函[7]。為了分析乙烯分子在催化劑表面的吸附,建立2 × 2的CuO/Cu2O(100)和CuO/Cu2O(111)的超胞來分別模擬立方體和八面體CuO/Cu2O納米晶晶面結構。2 × 2 × 1k點網格用于描述結構優化的布里淵區。網格的能量截斷值設為150 Ry,結構優化的力收斂標準低于0.05 eV·?-1(1 ? = 0.1 nm)。分子在催化劑表面上的吸附能Ead定義為:Ead=Etotal-Esurf-Emol,其中Etotal、Esurf和Emol分別代表吸附體系的總能量、干凈催化劑表面的能量和吸附分子在氣相中的能量。量化計算軟硬件詳細信息請見表4。
1.2 試劑和材料
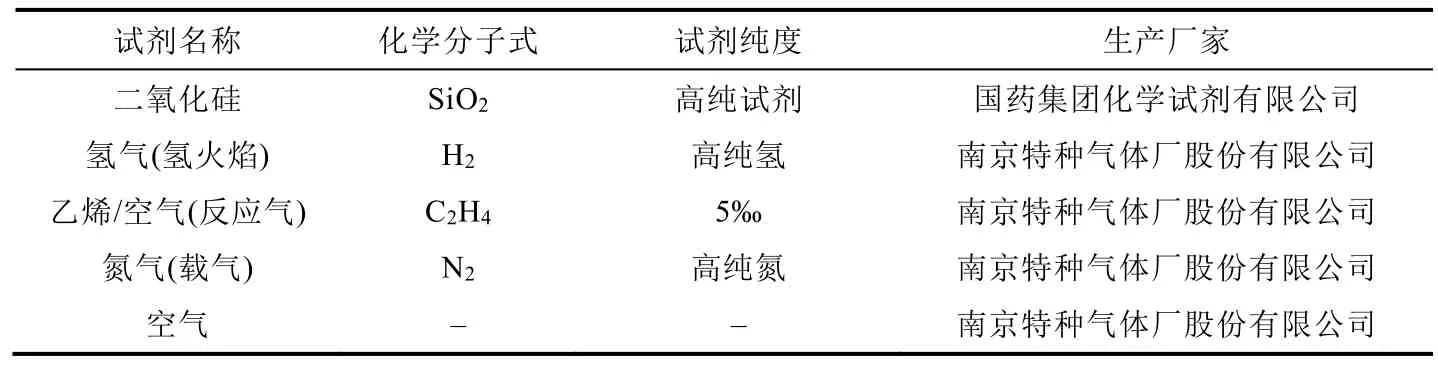
表1 實驗試劑
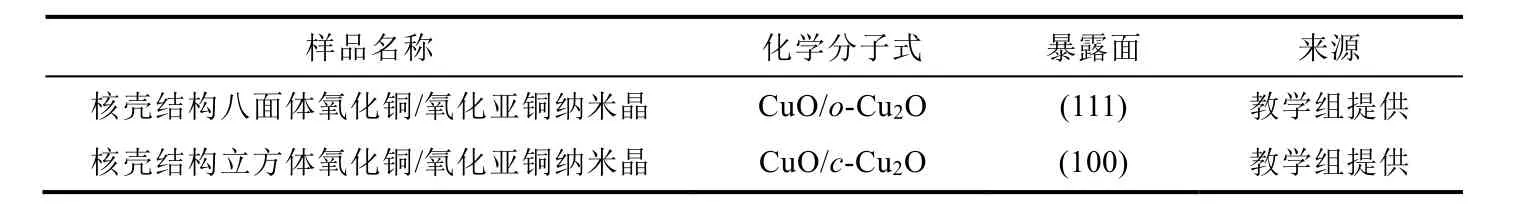
表2 催化劑信息
1.3 儀器和表征方法
1.3.1 催化劑活性測試

表3 實驗儀器
1.3.2 催化劑表征儀器
JSM-6700F型場發射掃描電子顯微鏡(日本電子公司),大功率轉靶X射線衍射儀(日本理學公司),ESCALAB 250型高性能電子能譜儀(Thermo-VG Scientific公司),JEM ARM-200F型高分辨透射電子顯微鏡(日本電子公司)。
1.3.3 量化計算軟硬件

表4 量化計算軟硬件
1.4 實驗步驟和方法
1.4.1 催化劑表征結果學習
教學組提供的兩組催化劑樣品是通過參考文獻[8]中方法合成得到,首先制備形貌規整的立方體與八面體Cu2O納米晶,表面經過氧化重構后形成了一層CuO薄膜,最終得到具有核殼結構的立方體和八面體CuO/Cu2O納米晶,分別記為CuO/c-Cu2O和CuO/o-Cu2O。該催化劑的相關表征結果會在講義中給出,表征結果分析作為實驗預習考核的一部分內容。
1.4.2 催化劑活性測試
在活性測試實驗中,小組同學得到兩組催化劑,分別是立方體納米晶,記為CuO/c-Cu2O,和八面體納米晶,記為CuO/o-Cu2O。催化劑的活性測試是在固定床式反應器上進行的,稱取50 mg催化劑與50 mg SiO2均勻混合后轉移進石英管中,反應氣體為0.5%乙烯-空氣混氣,氣體流速為20 mL·min-1,通過小組討論,選擇一系列溫度梯度進行測試,每個溫度下保溫15 min后測量。
1.4.3 量化計算
1.4.3.1 建立結構模型
使用Materials Studio 8.0軟件建立分子、催化劑表面和催化劑表面分子吸附的初始構型,包括建立乙烯分子模型;參考Huang等[2,3]報道的CuO/Cu2O界面模型,建立CuO/Cu2O(100)和CuO/ Cu2O(111)表面slab模型(圖1)來分別模擬CuO/c-Cu2O和CuO/o-Cu2O;在提供的CuO/Cu2O(111)和CuO/Cu2O(100)表面(已結構優化)上初步確定可能的吸附位點并建立分子的吸附結構。

圖1 通過Materials Studio 8.0軟件建立CuO/Cu2O(100) (圖A)和CuO/Cu2O(111) (圖B)的表面結構模型
其中,在建立2 × 2的CuO/Cu2O(100)和CuO/Cu2O(111)的超胞模型時,選擇x和y方向平行于表面而z方向垂直表面,同時在z方向選擇18 ?的真空層以避免表面之間的相互作用力。在CuO/Cu2O(100)和CuO/Cu2O(111)結構中,分別共有13和15層原子層(z坐標相同的為同一層),其中在頂端的四層中均添加O原子構成Cu和O原子個數比為1 : 1的CuO層,同時在量化計算時,均需固定底端的原子,固定層數分別為5和6層。
1.4.3.2 量化計算及查看結果
使用SIESTA軟件計算時,需準備輸入文件(后綴名為.fdf)和贗勢文件(后綴名為.psf)。準備輸入文件時,從Materials Studio 8.0軟件中導出小組分配好的結構文件(文件格式為.car),按格式生成計算所需要的坐標文件,設置計算方法和精度。在Xshell軟件操作界面下向超算集群提交計算任務。計算結束后,從輸出文件(OUT)中查看計算體系的能量值,計算分子在催化劑表面的吸附能。將得到的結構文件(后綴名為.STRUCT_OUT)轉化成Materials Studio 8.0軟件可識別的坐標文件格式,導入Materials Studio 8.0軟件中,在可視化界面下查看結構信息。
2 結果與討論
2.1 催化劑表征結果學習
2.1.1 Cu2O表征結果
c-Cu2O和o-Cu2O的FESEM與HRTEM表征結果如圖2所示。其中,立方體的平均尺寸約為600 nm,八面體的平均尺寸約為400 nm。根據HRTEM圖像圖2C和圖2F,c-Cu2O晶體的晶面間距為0.21 nm和0.21 nm,且晶面夾角約為90°,分別對應Cu2O的(020)面和(200)晶面(PDF卡片,No.78-2076)。o-Cu2O晶體的晶面間距為0.25 nm和0.25 nm,且晶面夾角約為71°,分別對應Cu2O的(111)面和(1-11)面(PDF卡片,No.78-2076)。以上可以說明制備得到的立方體和八面體兩種Cu2O納米晶主要暴露了(100)晶面和(111)晶面。

圖2 (A) c-Cu2O的FESEM圖;(B,C) c-Cu2O的HRTEM圖;(D) o-Cu2O的FESEM圖;(E,F) o-Cu2O的HRTEM圖
2.1.2 CuO/Cu2O表征結果
對表面重構后的催化劑樣品進行結構表征,結果如圖3所示。由HRTEM圖像可知,經過表面重構后,兩組納米晶表面產生一層約3 nm厚的薄膜。在XPS譜圖中Cu 2p3/2的結合能位于933.2 eV,同時在940-945 eV出現衛星峰,證明Cu2O納米晶表面的薄膜是被氧化產生的CuO薄膜。XRD圖譜在29.6°,36.4°,42.3°,61.4°,73.5°出現的衍射峰,分別對應Cu2O的(110),(111),(200),(220),(311)晶面(PDF卡片,No.78-2076),說明CuO/c-Cu2O與CuO/o-Cu2O的體相結構仍然保持為Cu2O晶相。

圖3 (A,B) CuO/c-Cu2O和CuO/o-Cu2O的HRTEM圖,(C,D) CuO/c-Cu2O和CuO/o-Cu2O的XPS譜圖和XRD譜圖
2.2 活性測試
活性測試實驗所得的結果如圖4所示。CuO/c-Cu2O在270 °C開始顯示活性,在300 °C時轉化率為21.5%;而CuO/o-Cu2O在220 °C就開始顯示活性,在300 °C轉化率高達80.3%,說明CuO/o-Cu2O對乙烯的催化活性比CuO/c-Cu2O高。對Arrhenius曲線進行擬合,CuO/c-Cu2O和CuO/o-Cu2O催化乙烯氧化的表觀活化能分別為117.0 ± 4.3 kJ·mol-1和77.7 ± 3.2 kJ·mol-1,兩者的表觀活化能差別很大,這說明兩種催化劑在反應中的反應活性位點不同,并且遵循不同的反應機理。
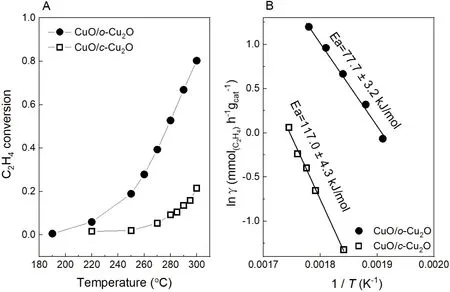
圖4 CuO/c-Cu2O和CuO/o-Cu2O催化乙烯氧化的轉化率(A)和Arrhenius曲線(B)
轉化率結果對于溫度穩定時間、反應氣流量十分敏感,且這兩個變量引起誤差無法消除,故學生在實驗過程中需保證達到溫度穩定時間并監控氣體流量確保其狀態穩定。在實驗過程中,反應器到達設定溫度后穩定15-20 min后開始測試,反應氣流量需控制在20 mL·min-1。
2.3 量化結果分析
催化劑表面上未飽和配位的原子往往作為活性中心,在CuO/Cu2O(100)表面,暴露的表面未飽和配位的原子為單一的O2c;而在CuO/Cu2O(111)表面,表面暴露的未飽和配位的原子為Cu3c和O3c(圖5(A)和(B))。計算乙烯在兩個表面上可能的吸附結構,確定CuO/Cu2O(100)上的化學吸附位點是(O2c,O2c),而CuO/Cu2O(111)上有兩種化學吸附的位點,一種是(Cu3c,Cu3c)位點,另一種是(O3c,O3c)位點(圖5(C)-(E))。

圖5 (A,B) CuO/Cu2O(100)和CuO/Cu2O(111)表面上乙烯的吸附位點;(C,D、E) 乙烯在CuO/Cu2O(100)和CuO/Cu2O(111)表面上穩定的吸附結構、吸附能(單位:eV)和C=C鍵長(單位:?)
乙烯分子在CuO/Cu2O(100)表面上的吸附作用強,吸附能為-6.21 eV,其中,C=C鍵由原來的1.338 ?伸長至1.527 ?。而在CuO/Cu2O(111)表面上,乙烯分子在(Cu3c,Cu3c)和(O3c,O3c)位點上的吸附能分別為-3.11和-2.34 eV,對應的C=C鍵長分別為1.459和1.527 ?。可見,CuO/Cu2O(111)表面上,(Cu3c,Cu3c)位點更易吸附乙烯分子,而(O3c,O3c)對C=C鍵活化作用更強。比較乙烯分子在兩個表面的吸附能,發現乙烯在CuO/Cu2O(100)表面吸附作用明顯比在CuO/Cu2O(111)表面上高。然而,根據Balandin火山曲線規律,過強或者過弱的吸附強度都會導致催化劑的催化活性下降,具有適中的吸附強度對催化反應最有利[9]。通過文獻學習可以推測在氣固催化反應中,這個“適中”的吸附強度對應的吸附能在-1 - -2 eV[10]。所以,CuO/Cu2O(100)表面對乙烯的吸附能過高,可能會對反應過程產生抑制作用,而CuO/Cu2O(111)表面上乙烯的吸附能適中,有利于進一步反應。
2.4 綜合分析
將活性測試實驗結果與量化計算結果進行匯總,見表5。活性測試實驗顯示,CuO/o-Cu2O對乙烯氧化反應的催化活性比CuO/c-Cu2O要高,通過轉化率數據計算得到兩者的表觀活化能差別很大,分別為77.7 ± 3.2 kJ·mol-1和117.0 ± 4.3 kJ·mol-1,說明這兩種催化劑表面暴露的活性位點不同,且CuO/o-Cu2O表面的催化活性優于CuO/c-Cu2O表面。基于催化劑表面的實驗表征結果,構建催化劑表面結構模型,能清晰直觀地看到CuO/o-Cu2O的活性位點為(Cu3c,Cu3c)或(O3c,O3c),而CuO/c-Cu2O的活性位點為(O2c,O2c)。相比于CuO/c-Cu2O(100)表面,乙烯分子在CuO/o-Cu2O(111)表面的吸附能比較適中,有利于反應的進行和產物的脫附[9,10],從而對應并解釋了活性測試實驗結果。

表5 實驗與量化計算結果
在實驗中引入量化計算,不僅是讓學生掌握一項實驗技能,更是讓學生具備實驗結合理論、宏觀聯系微觀的思維方式。然而,合理地將量化計算結果與實驗結果相對應是分析的重點與難點。尤其對于多相催化這種復雜的反應體系,由于量子化學計算的一些局限性,一般的計算結果只能部分反映體系的真實狀態。因此通過計算結果對實驗結果給出一個恰當的解釋是一個充滿挑戰的過程,往往需要查閱相關資料并結合體系自身的特點進行大量的討論,而通過這一步的練習,充分鍛煉了學生針對問題查閱文獻、討論思辨的能力,讓學生在這個過程中加深對催化劑結構與性能之間關系的理解。
3 結語
催化劑的催化性能與其表面結構息息相關。基于科研工作者提出的模型催化理念,實驗結合量化計算對表面催化過程進行研究,已成為研究催化劑表面科學的一種新方法。教學組將這一學科前沿研究方法引入到實驗教學中,填補了催化劑表面結構與催化性能關系相關實驗內容上的空白。通過文獻調研、實驗嘗試和驗證,教學組選擇理化性能穩定,形貌規整,暴露特定晶面的CuO/Cu2O納米晶作為模型催化劑,選擇綠色安全的乙烯氣體作為反應氣,成功地將該體系引入實驗教學。
教學中采取了實驗與量化計算相結合的方式。實驗中,學生進行活性測試并計算得到表觀活化能。隨后對乙烯分子在CuO/c-Cu2O和CuO/o-Cu2O表面吸附的結構進行量化計算,得到相應的結構信息和吸附能。量化計算結果與實驗結果相互驗證,互為解釋,共同構建對催化劑表面結構與催化性能關系的合理的解釋。通過對這個模型催化體系的學習,讓學生對物化教材上的相關知識有更深刻的理解,掌握了催化活性測試的實驗操作、表面結構模型的搭建方法和量化計算上機操作流程,鍛煉了他們的文獻調研能力,并激發高年級本科生的研究興趣、培養綜合實驗技能,為他們能夠順利進入研究生學習階段奠定良好基礎。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
