以聽力下降為首發癥狀的MELAS綜合征1例
2022-12-17 08:07:52葛小旭楊海鋒
醫學理論與實踐 2022年23期
關鍵詞:癥狀
葛小旭 楊海鋒 顧 俊
江蘇省如東縣人民醫院神經內科 226400
線粒體腦肌病伴乳酸中毒和卒中樣發作綜合征(Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes,MELAS)是一種母系遺傳疾病[1]。它由線粒體DNA突變引起,進而引發線粒體功能障礙導致的多系統代謝性疾病[2]。MELAS以卒中樣發作、癲癇發作、認知與精神障礙、高乳酸血癥、肌肉疲勞無力為主要臨床表現。臨床癥狀和實驗室檢查對診斷有重要幫助,確診主要依靠肌肉活檢和基因檢測[3]。目前,已報告超過30個線粒體DNA(mtDNA)基因位點突變可導致MELAS。最常見的基因突變是位點m.3243A>G,占MELAS病例的80%以上[3-4]。本文報道了1例在我院確診為MELAS的患者,觀察其臨床表現、影像學改變及預后情況。
1 病例資料
1.1 病史 患者男性,42歲,工人,因“突發言語不清1d”于2021年6月首次入院。主要表現為言語含糊不清,無肢體麻木無力,無肢體抽搐、意識障礙。既往史顯示患者30余歲時出現雙耳聽力下降,未正規治療,無其余特殊病史。已婚,育有1女,配偶及女兒皆體健。父親體健,其母患有“2型糖尿病”10余年。家族中姨媽患有糖尿病。
1.2 體格檢查 生命體征平穩,體型偏瘦,身高165cm,體重50kg,言語含糊,反應遲鈍,定向力、記憶力、計算力減退,雙側瞳孔等大等圓,直徑約3.0mm,對光反射靈敏,雙眼球活動正常,無凝視及眼震,聽力檢查不配合,伸舌居中,運動、感覺、共濟、腱反射檢查均無異常,雙下肢病理征陰性,頸軟無抵抗。
1.3 輔助檢查 血生化、C反應蛋白等檢驗未見異常,心電圖、心臟彩超、頸部血管超聲、頭顱CT平掃無異常。入院后初步診斷為腦梗死。入院當天患者突發肢體抽搐,雙眼上翻,呼之不應,約1min后自行停止。當時考慮腦炎可能,予完善腰椎穿刺術,外送自身免疫腦炎標本,結果回報腦脊液乳酸 7.3mmol/L,自身免疫性腦炎12項未見異常。頭顱MRI提示:左側顳頂枕葉見腦回樣異常信號,T1WI呈低信號,T2WI呈高信號,DWI呈高信號(見圖1)。患者空腹乳酸:乳酸 5.00mmol/L,運動5min后乳酸7.4mmol/L,休息10min后乳酸7.1mmol/L,此為乳酸簡易運動試驗,患者靜息狀態乳酸明顯高于正常,運動后乳酸升高,休息10min后未能回到乳酸基線水平,提示乳酸運動試驗陽性。考慮患者線粒體腦肌病可能,隨后完善頭顱MRS及基因檢測。頭顱MRS示NAA、Cho峰降低,出現倒置Lac峰(見圖2)。基因測序結果顯示:患者及其母親MT-TL1基因上均發生變異,為m.3243A>G(見圖3)。患者最終診斷為MELAS綜合征。
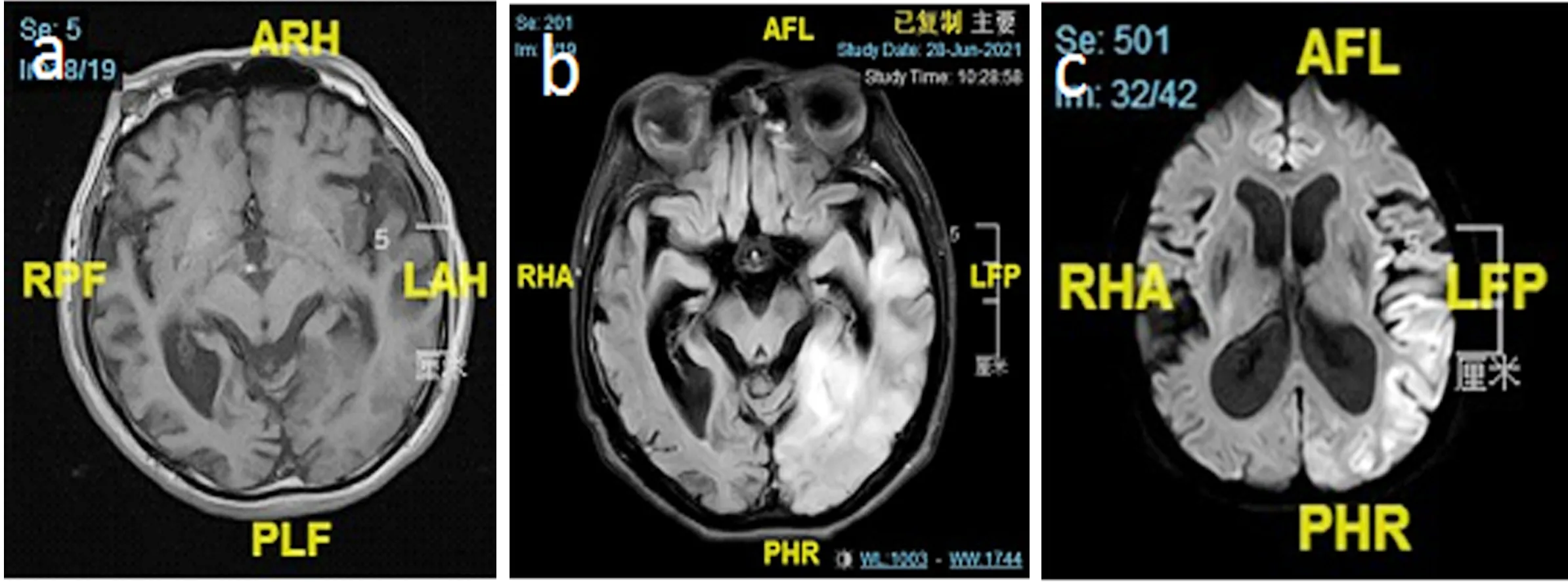
圖1 患者頭顱MRI
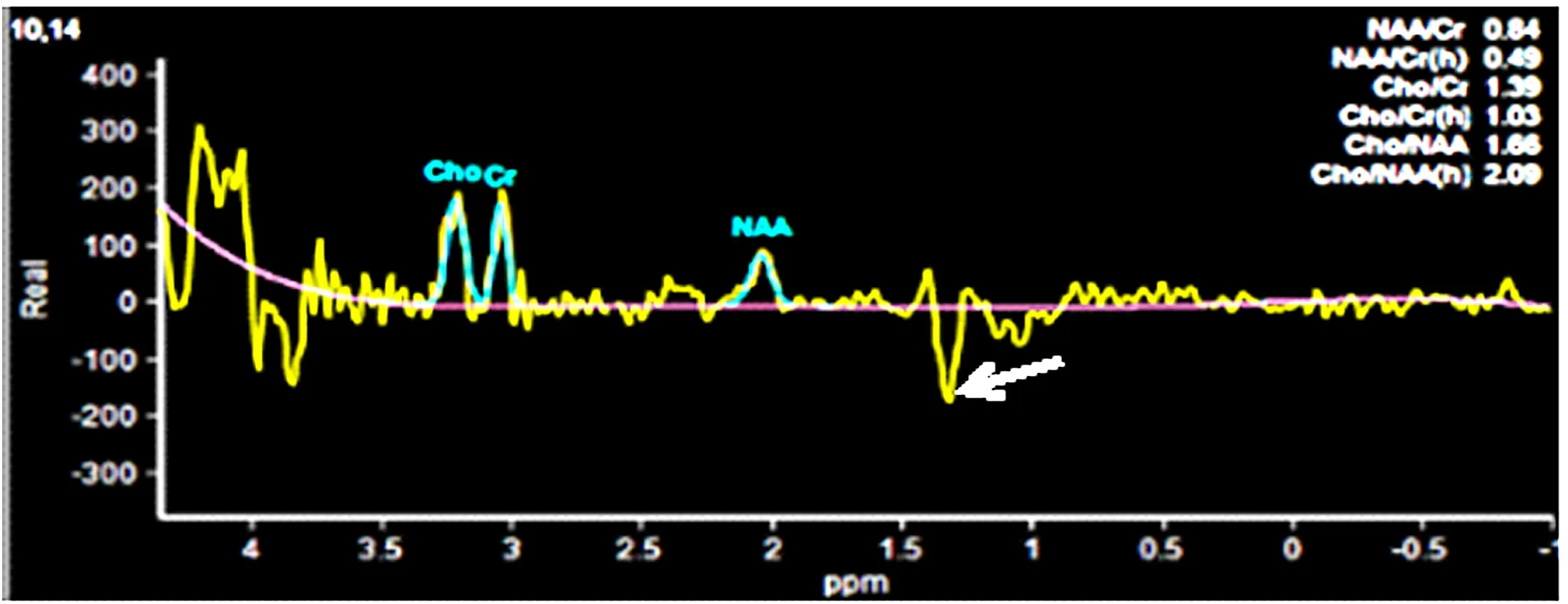
圖2 患者頭顱MRS
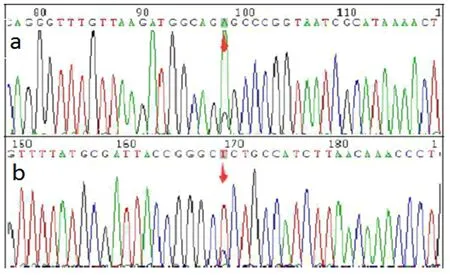
圖3 患者及其母親基因測序結果
1.4 治療與隨訪 入院后予L-精氨酸、艾地苯醌、輔酶Q10、左乙拉西坦患者癥狀好轉出院。2021年10月,患者因“認知功能下降4個月余”再次入院治療,頭顱MRI:左側顳頂枕葉,右側枕頂葉異常信號(見圖4)。聲阻抗報告:雙耳鼓室圖A型曲線,雙耳鐙骨肌反射全部未引出,雙耳鼓室功能正常。入院后仍給予L-精氨酸、輔酶Q10、艾地苯醌、多奈哌齊、左乙拉西坦等對癥治療后緩解出院。
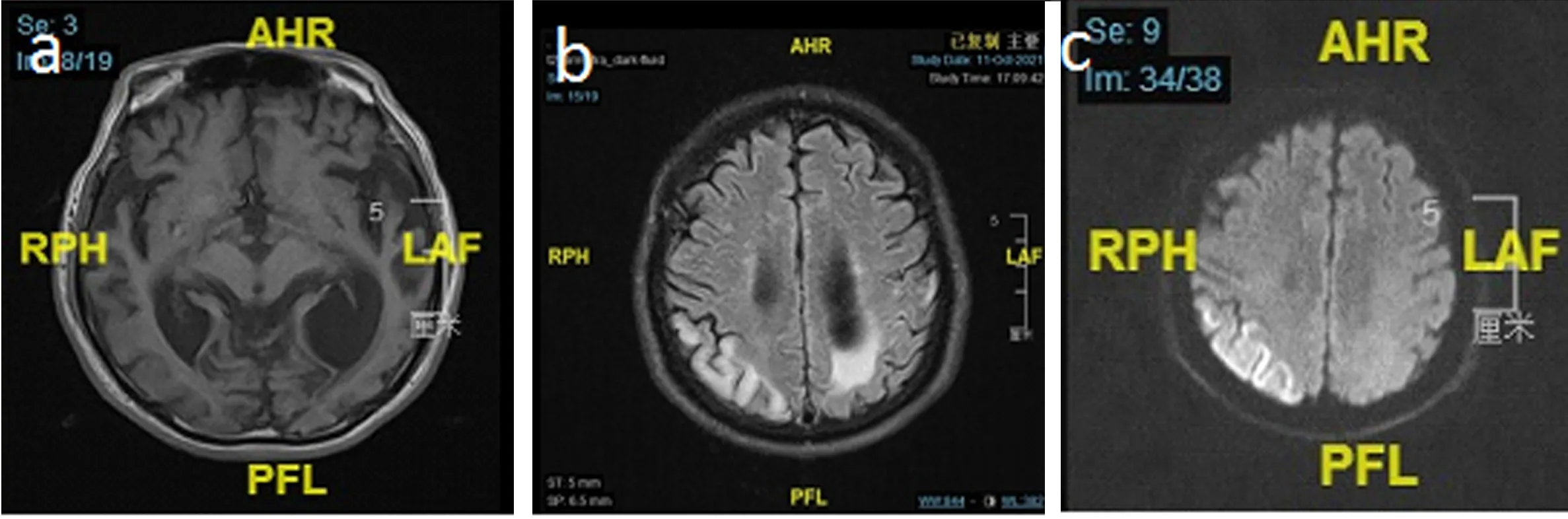
圖4 患者頭顱MRI
2 討論
MELAS綜合征是最常見的線粒體腦肌病類型,是一種母系遺傳的多系統代謝性疾病,男性發病率稍高于女性[1]。MELAS發病年齡多在40歲左右,發病機制與一氧化氮的缺乏有關,常見癥狀為卒中樣發作,出現偏癱、偏盲、失語、意識障礙等多種急性神經功能障礙,但大多數癥狀為一過性,不遺留后遺癥[5]。其他的癥狀包括癲癇、癡呆、頭痛,少見有耳聾、運動不耐受、智力低下、糖尿病和反復嘔吐[6-7]。生化檢測中可見腦脊液及血清乳酸值升高,但易受其他因素影響,不具有特異性[8]。
目前,MELAS診斷主要依靠臨床癥狀與影像學表現,肌肉活檢或基因檢查可確診[9]。頭顱MRI可見病灶位于皮質和皮質下,呈長T1、長T2異常信號,枕葉和顳葉最容易受累,病灶不符合顱內解剖血管分布。此外病灶具有進展性、可逆性、多發性以及呈現“此消彼長”的“游走性”特點[2]。患者第二次入院時頭顱MRI出現右側枕頂葉異常信號,符合病灶特點。MRS中出現乳酸雙峰對MELAS 診斷具有重要意義[10]。基因檢測中,最常見的是mtDNA3243A>G、13513G>A 及3271T>C 等變異位點。肌肉活檢典型病理改變是改良Gomori三色染色見不整紅邊纖維,琥珀酸脫氫酶染色見破碎藍染肌纖維。
本例患者入院時為卒中樣發作現象,后經過影像學及基因檢測證實為MELAS。但患者10年前雙耳就出現聽力下降,如以“一元論”解釋,考慮聽力下降為線粒體腦肌病所致。耳蝸中外毛細胞具有代謝活躍、高度的能量依賴性特點。而線粒體功能障礙會引起ATP水平下降,進而會造成供給外毛細胞功能下降,引起聽力下降[6]。此外,國內外也有以聽力下降為首發癥狀的MELAS報道。因此,筆者考慮患者首發癥狀為聽力下降。患者除了聽力下降,10年來未出現其他神經系統損害癥狀,病程進展緩慢,給早期診斷造成困難。
患者家系基因檢測中,患者及其母親均有MT-TL1基因發生m.3243A>G突變。而患者母親及姨媽均有糖尿病,線粒體功能缺失與糖尿病有密切關系。線粒體突變后,可影響胰島細胞氧化磷酸化功能,使胰島素合成與分泌減少[11]。線粒體mtDNA有閾值效應,即突變mtDNA達到一定比例,才導致疾病發生[12]。因此,其母是否存在線粒體腦肌病,需進一步行影像學檢查及肌肉活檢,但其母拒絕相關檢查。
MELAS綜合征的治療目前尚無有效特異方案。臨床主要是對癥治療緩解癥狀。補充L-精氨酸和輔酶Q10等,有助于改善患者能量代謝,提高生活質量。特別注意的是,一些藥物如苯巴比妥、雙胍類、他汀類等藥物可能影響線粒體功能,使用時需權衡利弊。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
