HPLC 法同時測定玉屏風口服液中12 種成分及質量一致性評價
2022-12-03 11:58:22超徐普劉柱施貝張文婷王峰
中成藥 2022年11期
岳 超徐普劉柱施貝 張文婷王峰
(浙江省食品藥品檢驗研究院, 浙江 杭州310052)
玉屏風為中醫扶正固表的經典名方,具有益氣、固表、止汗功效,主要用于表虛不固、自汗惡風、面色晄白或體虛易感風邪者,在新冠肺炎治療中作為推薦預防用藥,目前對該方成分的研究主要涉及皂苷類化合物,劑型大多為顆粒劑[1?4]、湯劑、散劑[3?10]。玉 屏風口服液收載于2020年版《中國藥典》中,大多以單一成分為質量控制指標,但該制劑制備工藝復雜,故上述標準無法準確評價其質量一致性。
本實驗以玉屏風口服液為研究對象,考察含量高、易提取并具有調節免疫力、解熱、抗炎、鎮痛等作用[11?18]的成分,最終選取黃芪中的毛蕊異黃酮葡萄糖苷、芒柄花素、槲皮素、毛蕊異黃酮,防風中的麻素苷、升麻素、5?O?甲基維斯阿米醇苷、芒柄花素,白術中的白術內酯Ⅰ、Ⅱ、Ⅲ及蒼術酮,建立HPLC 法同時測定其含量,并考察17 批樣品的質量一致性,以期為更全面合理地評價和控制該制劑質量提供參考依據。
1 材料
1.1 儀器 Agilent 1260 型高效液相色譜儀,配置G1314F 型DAD 檢測器、G316A 型柱溫箱、G376E型自動進樣器、G312B 型二元泵(美國Agilent 公司);電子分析天平(萬分之一,瑞士梅特勒?托利多公司);KH5200DE 型超聲波清洗器(昆山禾創超聲儀器有限公司);高速離心機(美國賽默飛世爾科技公司)。
1.2 試劑與藥物 升麻素苷(純度96.2%,批號111522?201712)、毛蕊異黃酮葡萄糖苷(純度97.6%,批 號111920?201606)、升麻素(純度100%,批號111710?200602)、5?O?甲基維斯阿米醇(純度97.4%,批號111523?20181)、亥茅酚苷(純度100%,批號111714?200501)、槲皮素(純度98.1%,批 號100081?201509)、白術內酯Ⅰ(純度99.9%,批號111975?201501)、白術內酯Ⅱ(純度99.9%,批號111976?201501)、白術內酯Ⅲ(純度99.9%,批號111978?201501)、蒼術酮(純度98.0%,批號DST90607?107)對照品(中國食品藥品檢定研究院);毛蕊異黃酮對照品(純度100%,成都普思生物科技股份有限公司,批號PS010251);芒柄花素對照品(純度98.0%,成都德思特生物技術有限公司,批號DST91202?011)。黃芪(膜莢)(批號121462?201304)、黃芪(蒙古)(批號120974?201612)、防風(批號120947?201409)、白術(炒)(批號120925?201611)對照藥材(中國食品藥品檢定研究院)。乙腈為色譜純(德國Merck 公司);甲醇為分析純(國藥集團化學試劑有限公司);水為純化水。玉屏風口服液共17批,均購于線下或線上藥店,具體信息見表1。
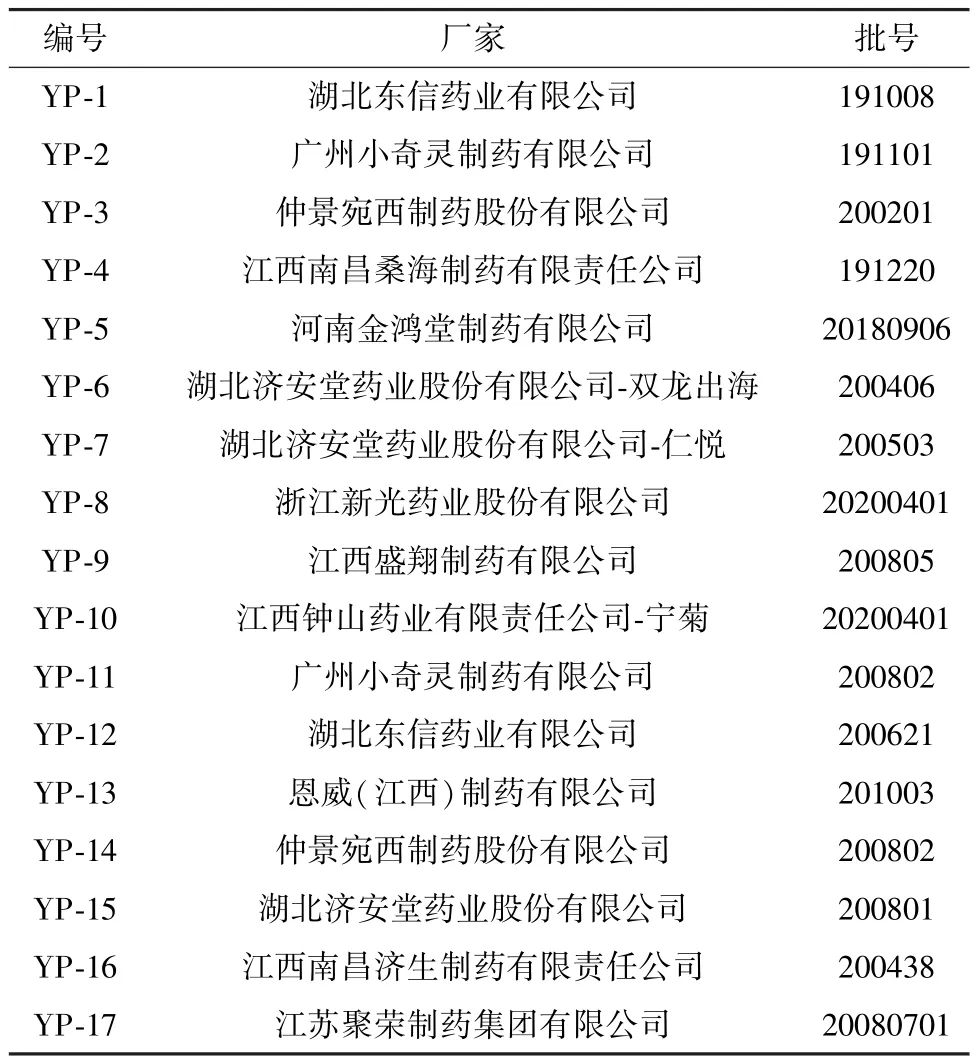
表1 樣品信息Tab.1 Information of samples
2 方法與結果
2.1 溶液制備
2.1.1 對照品溶液 精密稱取各對照品適量,甲醇稀釋成含升麻素苷1 067 μg/mL、毛蕊異黃酮葡萄糖苷1 019 μg/mL、升麻素1 246 μg/mL、5?O?甲基維斯阿米醇1 151 μg/mL、亥茅酚苷1 344 μg/mL、毛蕊異黃酮1 167 μg/mL、芒柄花素1 205 μg/mL、槲皮素1 185 μg/mL、白術內酯Ⅰ355 μg/mL、白術內酯Ⅱ350 μg/mL、白術內酯Ⅲ371 μg/mL、蒼術酮506 μg/mL 的溶液,分別吸取適量至同一量瓶中,甲醇稀釋,即得(各成分質量濃度范圍為35.01~67.20 μg/mL)。
2.1.2 供試品溶液 精密吸取本品1.0 mL 至20 mL量瓶中,80%甲醇定容至刻度,搖勻,靜置,取上清液,0.45 μm 有機濾膜過濾,即得。
2.1.3 陰性樣品溶液 分別制備缺黃芪、缺防風、缺炒白術的陰性樣品,按“2.1.2”項下方法制備,即得。
2.2 色譜條件 Waters SunFire C18色譜柱(250 mm×4.6 mm,5 μm);流動相水(A)?乙腈(B),梯度洗脫(0~32 min,9%~35% B;32~60 min,35%~80%B;60~65 min,80%~90%B;65~70 min,90% B;70~73 min,90%~9% B;73~75 min,9%B);體積流量1.0 mL/min;柱溫35 ℃;檢測波長300 nm(0~47 min,升麻素苷、升麻素、毛蕊異黃酮葡萄糖苷、5?O?甲基維斯阿米醇、毛蕊異黃酮、亥茅酚苷、槲皮素、芒柄花素)、220 nm(47~54 min,白術內酯Ⅲ)、275 nm(54~58 min,白術內酯Ⅱ)、220 nm(58~75 min,白術內酯Ⅰ、蒼術酮);進樣量10 μL。
2.3 系統適應性考察與專屬性試驗 精密吸取對照品、供試品、陰性樣品溶液各10 μL,在“2.2”項色譜條件下進樣測定,結果見圖1。由此可知,各成分均達到基線分離,分離度均大于1.5,陰性無干擾,表明該方法專屬性良好。
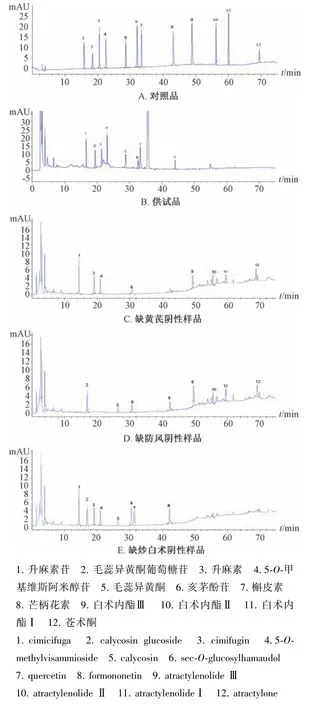
圖1 各成分HPLC 色譜圖Fig.1 HPLC chromatograms of various constitents
2.4 線性關系考察 分別精密吸取不同體積對照品溶液,在“2.2”項色譜條件下進樣測定。以對照品峰面積為縱坐標(Y),質量濃度為橫坐標(X)進行回歸,再將對照品溶液逐級稀釋,以信噪比(S/N)為3 時對應的質量濃度為檢出限,結果見表2,可知各成分在各自范圍內線性關系良好。

表2 各成分線性關系Tab.2 Linear relationships of various constituents
2.5 精密度、穩定性、重復性試驗
2.5.1 精密度 吸取同一份對照品溶液,在“2.2”項色譜條件下進樣測定6次,測得各成分峰面積RSD 分別為毛蕊異黃酮葡萄糖苷0.12%、亥茅酚苷0.24%、槲皮素0.09%、毛蕊異黃酮0.52%、升麻素苷0.32%、升麻素0.55%、5?O?甲基維斯阿米醇苷0.73%、芒柄花素0.48%、白術內酯Ⅰ1.41%、白術內酯Ⅱ1.24%、白術內酯Ⅲ1.06%、蒼術酮0.82%,表明儀器精密度良好。
2.5.2 穩定性 取同一份供試品溶液,于0、6、12、20、30、40、50 h 在“2.2”項色譜條件下進樣測定,測得各成分含量RSD 分別為毛蕊異黃酮葡萄糖苷0.33%、亥茅酚苷0.42%、槲皮素1.02%、毛蕊異黃酮0.84%、麻素苷1.07%、升麻素0.94%、5?O?甲基維斯阿米醇苷0.73%、芒柄花素0.84%、白術內酯Ⅰ1.11%、白術內酯Ⅱ1.13%、白術內酯Ⅲ0.94%、蒼術酮0.65%,表明溶液在50 h 內穩定性良好。
2.5.3 重復性 取同一批本品(編號YP?8),按“2.1.2”項下方法平行制備6 份供試品溶液,在“2.2”項色譜條件下進樣測定,測得各成分含量RSD 分別為毛蕊異黃酮葡萄糖苷0.59%、亥茅酚苷0.75%、槲皮素0.73%、毛蕊異黃酮0.94%、麻素苷1.26%、升麻素0.96%、5?O?甲基維斯阿米醇苷0.93%、芒柄花素0.97%、白術內酯Ⅰ0.76%、白術內酯Ⅱ1.34%、白術內酯Ⅲ1.29%、蒼術酮0.73%,表明該方法重復性良好。
2.6 加樣回收率試驗 取9 份各成分含量已知的本品(編號YP?8),每份0.50 mL,分別加入0.10、0.20、0.30 mL 對照品溶液各3份,按“2.1.2”項下方法制備供試品溶液,在“2.2”項色譜條件下進樣測定,計算回收率。結果,各成分平均加樣回收率(RSD)分別為毛蕊異黃酮葡萄糖苷97.6%(2.45%)、亥茅酚苷95.3%(2.87%)、槲皮素 94.7%(1.18%)、毛蕊異黃酮 97.6%(1.04%)、升麻素苷97.1%(1.60%)、升麻素97.1%(1.60%)、5?O?甲基維斯阿米醇苷100.7%(1.73%)、芒柄花素96.3%(1.99%)、白術內酯Ⅰ95.8%(2.56%)、白術內酯 Ⅱ 95.3%(2.79%)、白術內酯Ⅲ96.6%(3.07%)。
2.7 樣品含量測定 取17 批樣品,按“2.1.2”項下方法制備供試品溶液,在“2.2”項色譜條件下進樣測定,計算含量,結果見表3。由此可知,升麻素苷、毛蕊異黃酮苷、5?O?甲基維斯阿米醇苷、毛蕊異黃酮含量較高;不同廠家、批次樣品中各成分含量之間差異較大,如YP3 與YP7,并且相同廠家不同批次樣品亦然,如YP4 與YP16;所有批次樣品中均未檢出白術內酯Ⅰ。
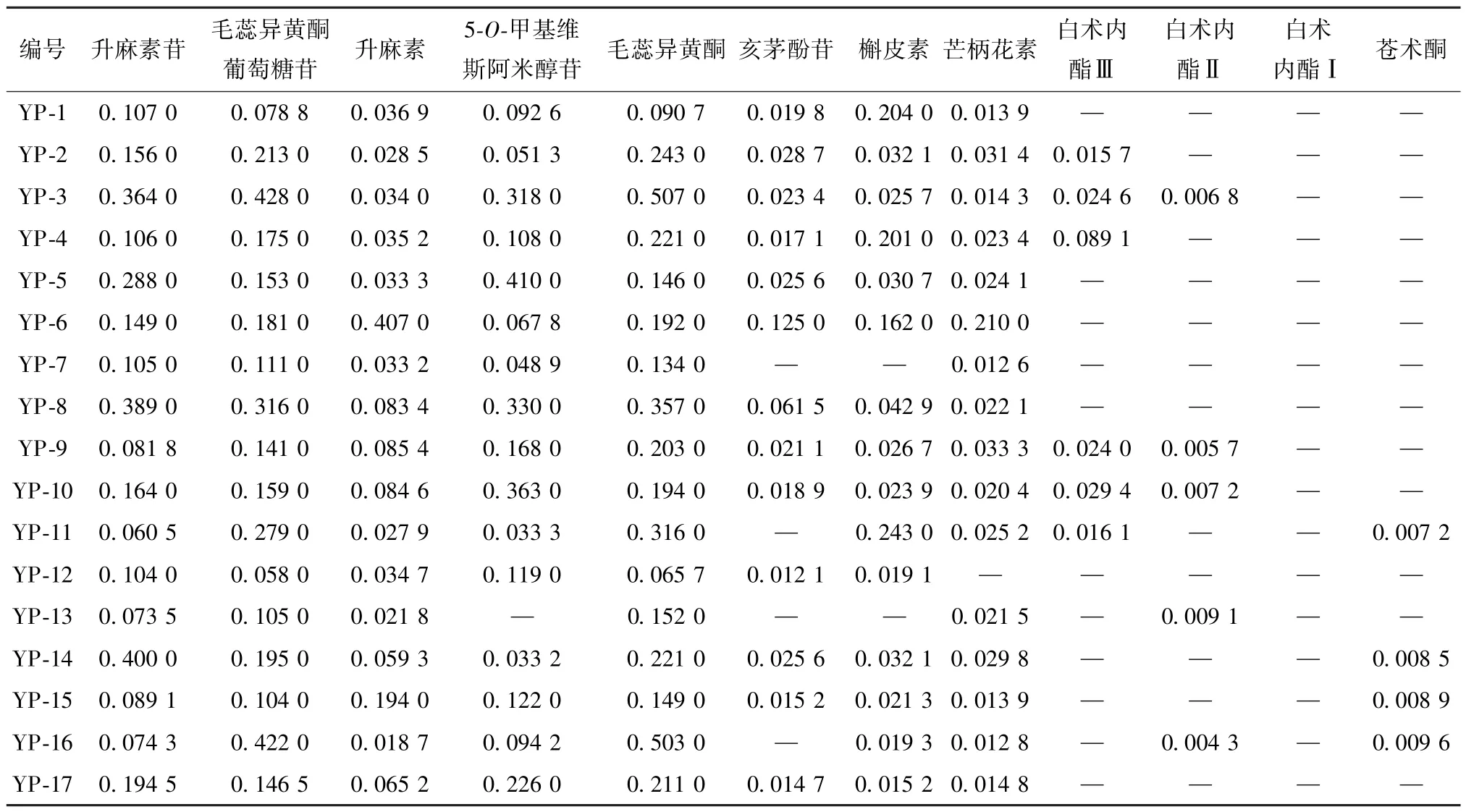
表3 各成分含量測定結果(mg/mL)Tab.3 Results of content determination of various constituents
2.8 化學模式識別
2.8.1 主成分分析 以表3 數據為變量,采用SPSS 22.0 軟件進行主成分分析,計算相關系數的特征值和方差貢獻率,提取特征值>1者,總方差貢獻率見表4,主成分圖分析見圖2,特征值載荷圖見圖3。由此可知,前4 個主成分特征值均>1,方差累積貢獻率達94.436%;樣品YP6、YP11、YP16 距離原點較遠,可明顯與其他樣品區分開;與原始變量關系最密切的目標物為芒柄花素、升麻素、亥茅酚苷、毛蕊異黃酮葡萄糖苷、毛蕊異黃酮,來自黃芪、防風藥材,對主成分的貢獻較大。
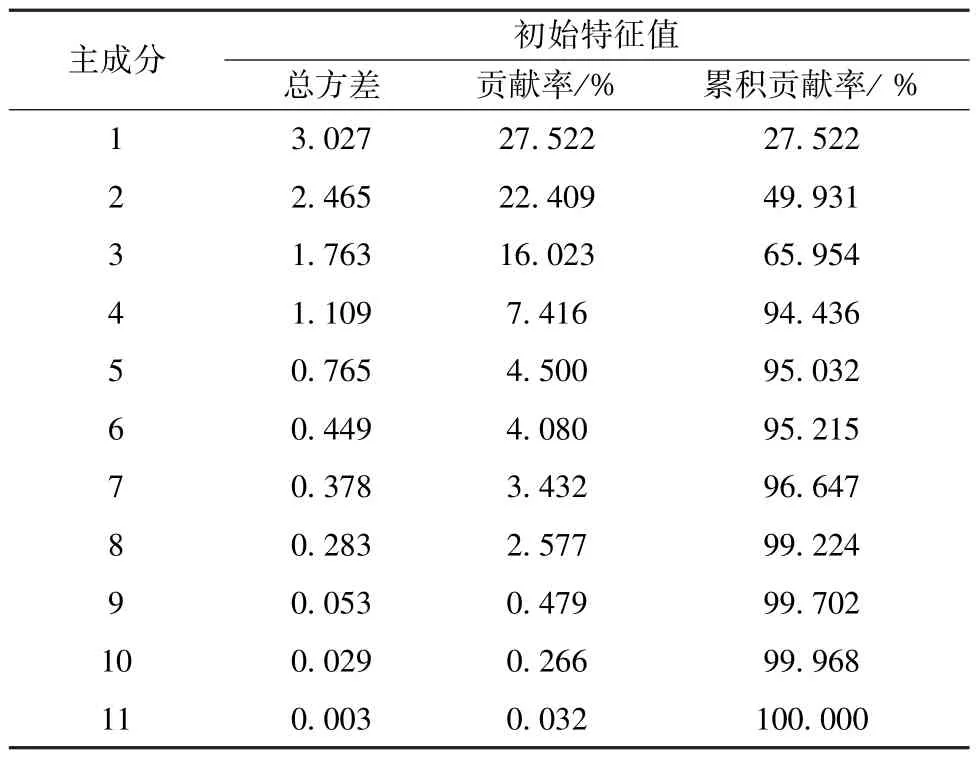
表4 主成分特征值的總方差貢獻率Tab.4 Total variance contribution rates of characteristic values for principal components
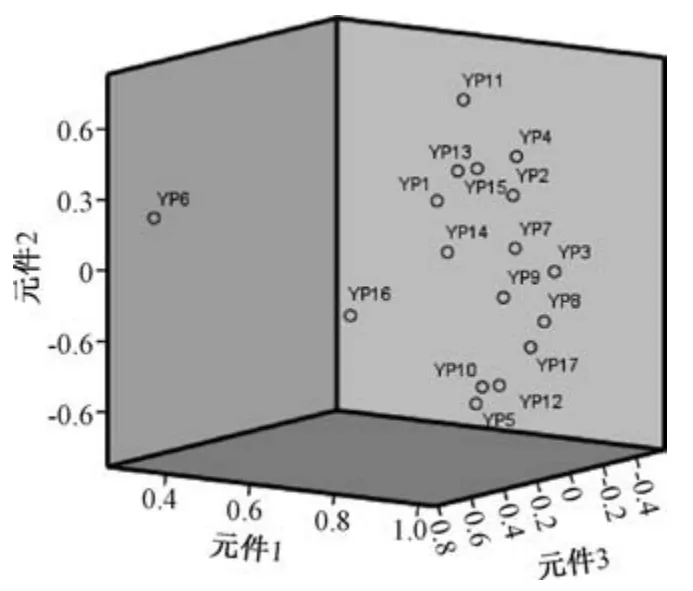
圖2 17 批樣品主成分分析圖Fig.2 Principal component analysis diagram for seventeen batches of simples
2.8.2 聚類分析 以表3 數據為變量,采用SPSS 22.0 軟件中的組間聯接系統聚類法,以平方歐式距離為測度進行聚類分析,結果見圖4。由此可知,17 批樣品聚為4類,YP3、YP8 為一類,YP6為一類,YP11、YP16 為一類,其余樣品為一類,與圖3 一致;YP3、YP8,YP11、YP16 中各成分含量占比相似,總和最高,與表3 一致,表示模型穩定可靠。
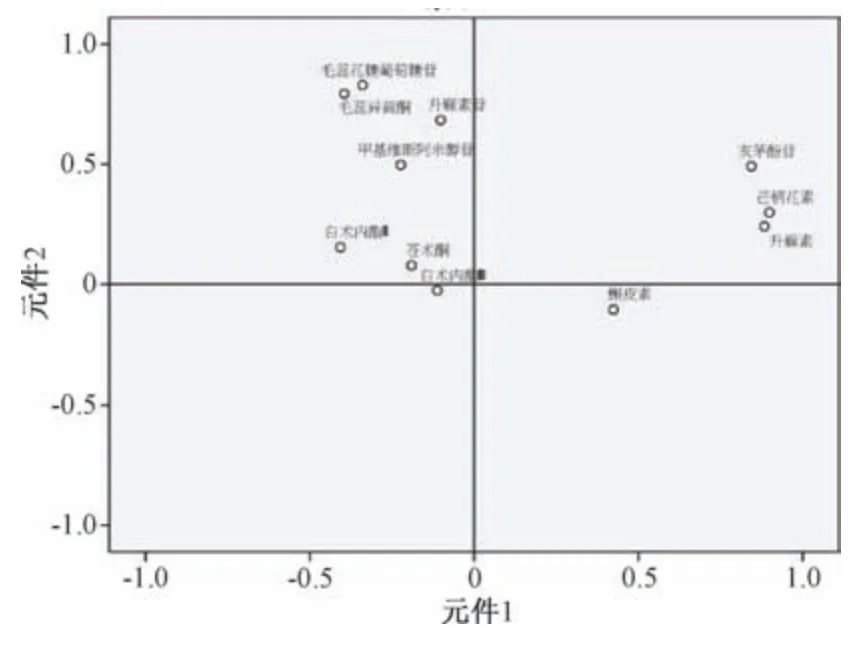
圖3 主成分特征值載荷圖Fig.3 Loading diagram for characteristic values of principal components
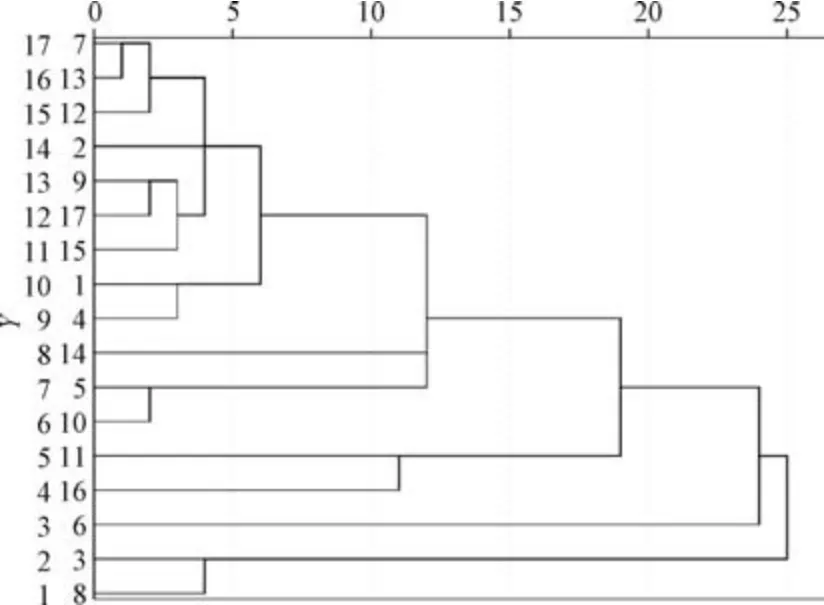
圖4 17 批樣品聚類樹狀圖Fig.4 Cluster dendrogram of seventeen batches of samples
3 討論
3.1 測定方法優化 本實驗分別考察了不同稀釋溶劑(甲醇、乙醇、水、乙腈、80%甲醇、50%甲醇),發現80%甲醇稀釋時雜質干擾少,各成分含量最高。再對供試品、對照品溶液進行全波長掃描,發現白術內酯Ⅱ在275 nm,白術內酯Ⅰ、白術內酯Ⅲ、蒼術酮在220 nm 處有最大吸收,其余成分在220、250、300 nm 處均有吸收。為了使各成分色譜峰的峰高協調統一,最終確定300 nm 作為主要檢測波長,并選擇“2.2”項下切換程序。
3.2 含量分析 不同批次玉屏風口服液中各成分含量差異較大,其原因可能來自生產工藝和原藥材,其中前者濃縮、醇沉淀、過濾等流程未作參數規定,主觀經驗依賴性強,個體差異大;后者產地、基源、生長方式、年限、用藥部位、炮制規格等因素均會影響質量一致性[19?23]。本實驗通過主成分分析得到5 個區分不同批次樣品的關鍵成分(芒柄花素、升麻素、亥茅酚苷、毛蕊異黃酮葡萄糖苷、毛蕊異黃酮),可更好地控制其質量一致性。
4 結論
本實驗建立HPLC 法同時測定玉屏風口服液中12 種成分的含量,并進行質量一致性評價,該方法簡便、可靠、準確,檢測指標全面,可為該制劑質量控制研究提供新思路,也能為提升其質量評價標準提供參考。
