柴胡屬飲片中藏柴胡摻偽檢測方法研究
2022-11-11 12:58:56趙丹彤高一軍畢天琛王素香鄭兆顯林永強
藥學研究 2022年10期
趙丹彤,高一軍,畢天琛,王素香,鄭兆顯*,林永強
(1.菏澤市食品藥品檢驗檢測研究院,山東 菏澤 274000;2.菏澤市立醫院,山東 菏澤 274031;3.山東省食品藥品檢驗研究院/山東省中藥標準創新與質量評價工程實驗室,山東 濟南 250101)
柴胡始載于《神農本草經》,列為上品,至《本草圖經》改名為柴胡,其性寒,味微苦,歸肝、膽經,具有疏散退熱、疏肝解郁之功效,主要用于感冒發熱、寒熱往來等疾病[1-3]。《中國藥典》2020年版(一部)收載柴胡為傘形科植物柴胡(BupleurumchinenseDC.)或狹葉柴胡(BupleurumscorzonerifoliumWilld.)的干燥根,分別習稱“北柴胡”和 “南柴胡”[1]。我國柴胡屬植物種類較多,分布廣泛,目前《中國植物志》記載有42種、17變種、7 變型[4-5]。市售柴胡藥材品種繁多,各省炮制規范中收錄有竹葉柴胡[6]、紅柴胡[7]等多個品種。因需求量較大且野生柴胡資源匱乏,目前藥用柴胡主要以栽培種植為主。由于柴胡屬植物的性狀較為接近,部分栽培品主要性狀特征不明顯,在采集和使用時難以鑒別,以致長期以來多種混偽品與正品柴胡混淆用藥的現象較為普遍。
藏柴胡為傘形科植物窄竹葉柴胡[BupleurummarginatumWall.ex DC.var.stenophyllum(Wolff.)Shan et Y.Li]的干燥根,為貴州等地方標準收載品種,主產于我國西南、甘肅等地區[8-9]。因其柴胡皂苷含量和產量顯著高于其他品種柴胡,且價格較低,目前市場上存在藏柴胡摻偽或混入其他柴胡屬飲片銷售、使用的情況[10]。由于藏柴胡與北柴胡等品種性狀相似,化學成分基本一致,常規薄層色譜法、液相色譜法等無法進行有效區分[11-14],且目前藏柴胡摻偽量也無準確計算方法。因此,本研究以Nepasaikosaponin K差異標志物為指標,采用高效液相色譜-串聯質譜技術(HPLC-MS/MS)技術,建立柴胡屬飲片中藏柴胡摻偽檢測方法,為柴胡屬飲片質量評價與監管提供有力的技術保障,同時為中藥摻偽檢測技術研究提供新的思路與方法。
1 儀器與材料
1.1 儀器 1260-6460高效液相色譜-三重四極桿質譜聯用儀,ESI離子源(美國Agilent 公司);XPR2型百萬分之一分析天平、AB104-S型萬分之一分析天平(瑞士Mettler Toledo公司);FRQ-1010T型超聲波清洗器(杭州法蘭特超聲波科技有限公司);FSJ-A05N6型粉碎機(小熊電器股份有限公司)。
1.2 材料 Nepasaikosaponin K對照品(CAS:405229-61-0,批號:DSTAC019202,純度≥98%)購自成都樂美天醫藥科技有限公司;甲醇、乙腈、甲酸均為色譜純;氨水為分析純;水為超純水。
21批柴胡屬飲片樣品信息見表1,其中S1~S21均經菏澤市食品藥品檢驗檢測研究院沙啟營主任中藥師鑒定為正品,S22~S24鑒定為藏柴胡摻偽北柴胡樣品。

表1 柴胡屬飲片樣品信息
2 方法與結果
2.1 溶液的制備
2.1.1 對照品溶液的制備 精密稱取Nepasaikosaponin K對照品5 mg,置10 mL量瓶中,以甲醇溶解并稀釋至刻度,即得對照品儲備液。取對照品儲備液適量于量瓶中,加甲醇稀釋至刻度,分別制成每1 mL含Nepasaikosaponin K 0.1、0.2、0.5、1.0、2.5、5.0、10.0、25.0、75.0 μg的系列工作對照品溶液。
2.1.2 供試品溶液制備 取柴胡屬飲片粉末(過四號篩)約0.5 g,精密稱定,置具塞錐形瓶中,精密加入含5%濃氨試液的甲醇溶液25 mL,密塞,稱定重量,30 ℃水溫超聲處理(功率500 W,頻率40 kHz)30 min,取出,放冷,再稱定重量,用含5%濃氨試液的甲醇溶液補足減失的重量,搖勻,用0.22 μm微孔濾膜濾過,即得。
2.2 檢測條件
2.2.1 色譜條件 色譜柱:Waters ACQUITY UPLC HSS T3 色譜柱(2.1 mm ×100 mm,1.8 μm);柱溫40 ℃;流速0.25 mL·min-1;進樣量5 μL;以0.1%甲酸水溶液(A)-乙腈(含0.1%甲酸)(B)為流動相,梯度洗脫程序:0→2 min,70% A;2→5 min,70% A→55% A;5→8 min,55% A;8→9 min,55% A→70% A;9→10 min,70% A。
2.2.2 質譜條件 采用質譜檢測器,電噴霧負離子(ESI-)進行多反應監測(MRM),選擇m/z 943.6→635.5、m/z 943.6→781.5、m/z 943.6→797.4為檢測離子對進行檢測。干燥氣溫度:300 ℃;霧化器壓力:20 Psi;干燥氣流速:10 L·min-1;鞘流氣溫度:350 ℃;鞘流氣流速:10 L·min-1;毛細管電壓:3 500 V。檢測離子對質譜參數見表2。Nepasaikosaponin K的 HPLC-MS/MS一級、二級質譜圖見圖1。Nepasaikosaponin K對照品、各柴胡屬飲片及藏柴胡摻偽飲片的總離子流圖和選擇離子對色譜圖見圖2。

圖1 Nepasaikosaponin K的一級質譜圖(A)及二級質譜圖(B)
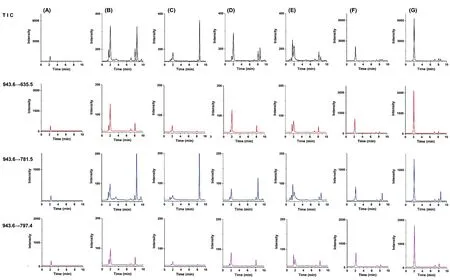
A.Nepasaikosaponin K對照品;B.北柴胡;C.南柴胡;D.竹葉柴胡;E.錐葉柴胡;F.摻偽50%藏柴胡的北柴胡;G.藏柴胡
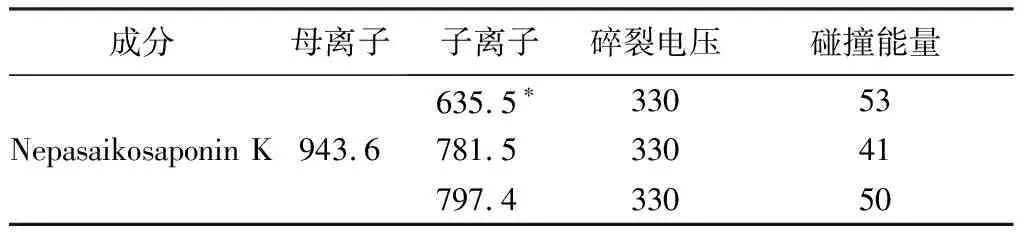
表2 檢測離子對質譜參數
取“2.1.1”項下1.0 μg·mL-1工作對照品溶液,進樣量為5 μL,Nepasaikosaponin K的色譜峰(m/z 943.6→635.5)信噪比應大于10∶1。
2.3 方法學考察
2.3.1 線性關系考察 分別取系列工作對照品溶液5 μL,按“2.2”項下檢測條件進行測定。以對照品濃度(μg·mL-1)為橫坐標(X),特征定量離子對943.6→635.5峰面積為縱坐標(Y),繪制標準曲線,計算回歸方程為Y=3 113.2X+19.084,R2=0.998 6。表明Nepasaikosaponin K質量濃度在0.1 μg·mL-1~75.0 μg·mL-1的范圍內線性關系良好。
2.3.2 精密度試驗 取“2.1.1”項下1.0 μg·mL-1工作對照品溶液,按“2.2”項下檢測條件連續進樣6次,記錄峰面積。結果表明Nepasaikosaponin K特征定量離子對943.6→635.5峰面積的RSD值為1.5%,表明本方法精密度良好。
2.3.3 重復性試驗 精密稱取同一批藏柴胡飲片6份(編號:S14),按“2.1.2”項下方法制備供試品溶液,按“2.2”項下檢測條件進行測定,記錄峰面積,計算得到Nepasaikosaponin K的平均含量為483.1 μg·g-1,RSD值為1.9%,表明本方法重復性良好。
2.3.4 穩定性試驗 取同一藏柴胡飲片供試品溶液,按“2.2”項下檢測條件分別于0、2、4、6、8、10、12、24 h進行測定,記錄峰面積,結果表明Nepasaikosaponin K特征定量離子對943.6→635.5峰面積的RSD值為1.7%,表明供試品溶液在24 h內穩定性良好。
2.3.5 加樣回收率試驗 精密稱取已知Nepasaikosaponin K含量的北柴胡飲片供試品9份,每份約0.5 g,分別加入工作對照品溶液,按“2.1.2”項下方法制備供試品溶液,按“2.2”項下檢測條件進行測定,記錄峰面積,計算加樣回收率及RSD值,結果見表3,表明方法的準確度良好。

表3 北柴胡飲片中Nepasaikosaponin K加樣回收率試驗結果
2.3.6 檢測限與定量限考察 取北柴胡飲片供試品溶液(編號:S3,Nepasaikosaponin K含量為16.56 μg·g-1),適當稀釋后按“2.2”項下檢測條件進行測定,結果表明含量水平為0.55 μg·g-1時Nepasaikosaponin K定量、定性離子信噪比均大于3,含量水平為1.66 μg·g-1時Nepasaikosaponin K定量離子信噪比均大于10。綜合考慮樣品基質效應,擬定柴胡屬飲片中Nepasaikosaponin K的檢測限為0.6 μg·g-1,定量限為1.7 μg·g-1。
2.4 Nepasaikosaponin K在柴胡屬飲片中分布情況 分別取各柴胡屬飲片,照“2.1.2”項下方法制備供試品溶液,按“2.2”項下檢測條件進行測定,記錄峰面積并按外標法計算樣品中Nepasaikosaponin K的含量,每個樣品平行測定3次,結果見表4。
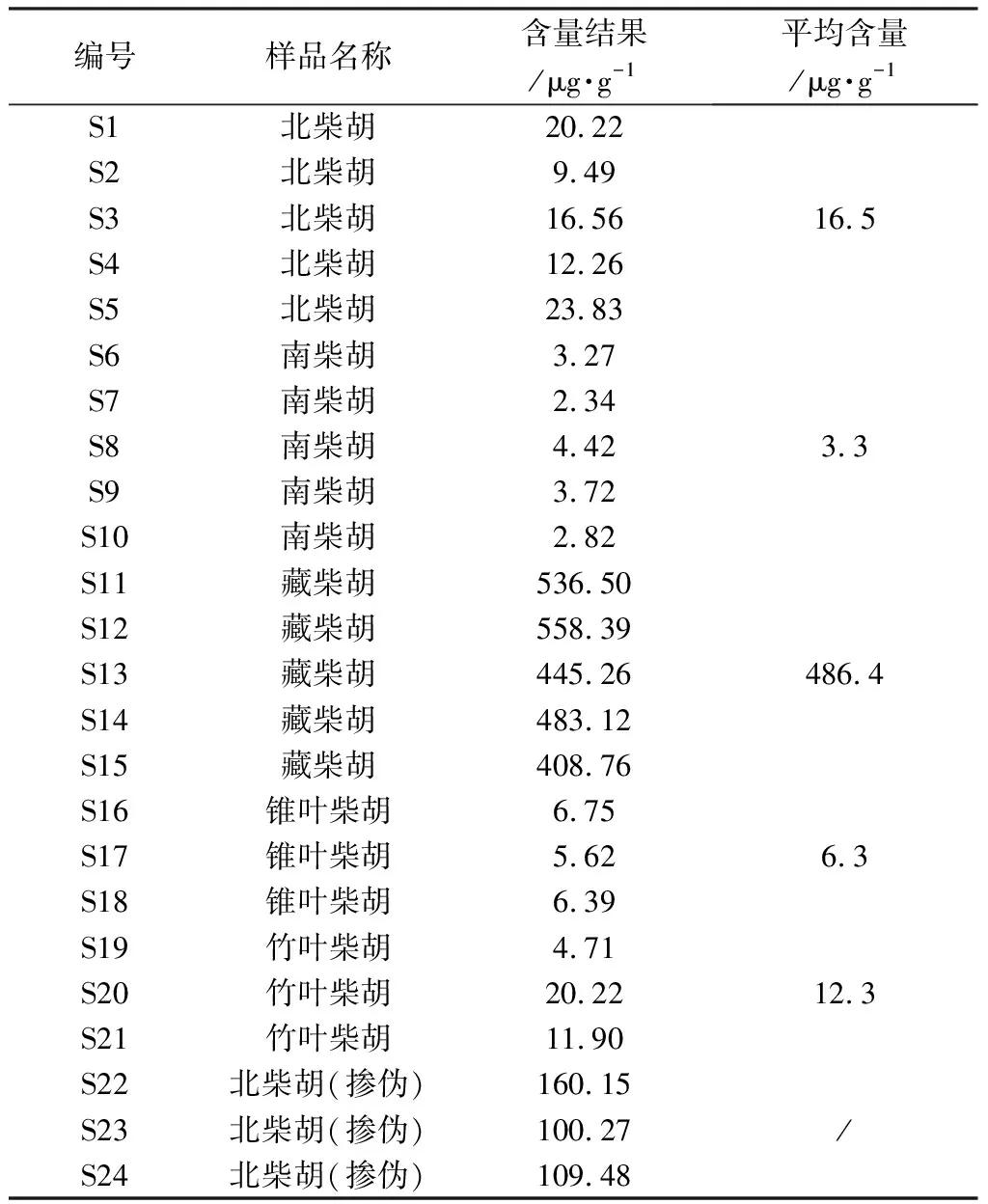
表4 柴胡屬飲片樣品Nepasaikosaponin K含量測定結果(n=3)
2.5 藏柴胡摻偽柴胡屬飲片檢測方法研究
2.5.1 藏柴胡摻偽柴胡屬飲片檢測方法線性關系考察 以北柴胡為例,將藏柴胡與同一份北柴胡樣品按比例混合,制成藏柴胡摻偽比例為0%、5%、10%、20%、40%、50%、70%、90%、100%的混合樣品9份,照“2.1.2”方法制備,照“2.2”項下檢測條件進行測定,以供試品溶液 m/z 943.6→635.5的提取離子流中Nepasaikosaponin K的色譜峰峰面積為縱坐標,藏柴胡摻偽北柴胡飲片的比例為橫坐標,繪制標準曲線。試驗結果表明,兩者呈良好的線性關系,線性回歸方程為:Y=28 681X+2 246.7,r=0.992;說明該方法在藏柴胡摻偽北柴胡飲片比例0~100%范圍內,可對北柴胡飲片中藏柴胡摻偽量進行定量檢測。
2.5.2 柴胡屬飲片藏柴胡摻偽比例測定方法研究 取各柴胡屬飲片,分別加入一定量藏柴胡飲片,制成藏柴胡摻偽比例為5%、20%、50%的混合樣品,照“2.1.2”項下方法制備供試品溶液,按“2.2”項下檢測條件進行測定,記錄峰面積并按外標法計算混合樣品中Nepasaikosaponin K的含量,照公式(1)計算藏柴胡摻偽比例。由表5結果可知,各柴胡屬飲片實際摻偽量與計算摻偽量平均偏差在0.6%~1.4%之間,說明該方法能準確測定柴胡屬飲片中藏柴胡摻偽比例。

表5 柴胡屬飲片實際摻偽量與計算摻偽量測定結果
公式(1)
其中P摻%為藏柴胡摻偽比例(%);CSSK混為藏柴胡摻偽柴胡屬飲片混合樣品中Nepasaikosaponin K的含量(μg·g-1);CSSK藏為藏柴胡樣品中Nepasaikosaponin K的含量(μg·g-1);CSSK樣為各柴胡屬飲片樣品中Nepasaikosaponin K的含量(μg·g-1)。
2.5.3 市售北柴胡飲片中藏柴胡摻偽比例測定 取市售疑似摻偽藏柴胡的北柴胡飲片,采用《中國藥典》2020年版(四部)< 0211 藥材和飲片取樣法 >中四分法取樣,并根據性狀鑒別特征分別挑選出少許北柴胡、藏柴胡,照“2.1.2”項下方法分別制備供試品溶液、藏柴胡供試品溶液及北柴胡供試品溶液,按“2.2”項下檢測條件進行測定,記錄峰面積并按外標法計算混合樣品中Nepasaikosaponin K的含量,照公式(1)計算藏柴胡摻偽比例,結果見表6。

表6 市售疑似摻偽藏柴胡的北柴胡飲片摻偽比例測定結果(n=3)
2.6 北柴胡飲片中藏柴胡摻偽擬定限度研究 根據市場調研及數據分析,參照《中國藥典》2020年版(四部)<0212 藥材和飲片檢定通則>所述雜質通常不得過3%的限度,同時考慮柴胡存在交叉種植、種質多樣及性狀易混淆的可能,擬定北柴胡飲片中藏柴胡摻偽限度為5%。
取北柴胡飲片樣品5批次,分別加入一定量藏柴胡飲片,制成藏柴胡摻偽比例為5%的混合樣品,照“2.1.2”項下方法制備供試品溶液,按“2.2”項下檢測條件進行測定,記錄m/z 943.6→635.5峰面積。結果如表7所示,按外標法計算混合樣品中Nepasaikosaponin K的平均含量為1.04 μg·mL-1。因此,本研究擬定Nepasaikosaponin K對照品溶液的限度為1 μg·mL-1。當北柴胡飲片供試品溶液的提取離子流色譜中,同時檢出與對照品溶液一致的特征離子對m/z 943.6 →635.5、m/z 943.6→797.4、m/z 943.5→781.5對應的色譜峰,且供試品色譜中943.6→635.5的色譜峰面積值大于1 μg·mL-1對照品溶液中相應的峰面積值時,視為北柴胡飲片中人為摻偽藏柴胡。

表7 不同批次藏柴胡摻偽北柴胡飲片Nepasaikosaponin K含量檢測結果(n=3)
3 討論
3.1 檢測條件優化
3.1.1 檢測目標物與方法選擇 柴胡皂苷為柴胡屬藥材主要活性成分之一,目前多采用液相色譜法對柴胡中環氧醚型(柴胡皂苷a、c、d)和異環雙烯型(柴胡皂苷b1、b2)等成分進行檢測[12,15];而其他柴胡皂苷成分研究較少,現有方法存在靈敏度低、各皂苷成分相互干擾的缺陷。Nepasaikosaponin K在北柴胡、南柴胡等品種中含量較低,而在藏柴胡中含量較高,有利于提高摻偽后目標成分檢測的靈敏度;但該皂苷又與多種皂苷結構相似,液相分離難度較大,故采用液質聯用法進行檢測,以提高靈敏度,滿足柴胡屬飲片中藏柴胡摻偽檢測方法準確度要求。
3.1.2 供試品溶液制備條件選擇 以含濃氨試液的甲醇溶液為提取溶劑,考察超聲時間(30、45、60 min)、溶劑體積(25、50、75 mL)及濃氨濃度(2%、5%、10%)對供試品溶液中定量特征離子對m/z 943.6→635.5色譜峰峰面積的影響。結果表明,兼顧時間及試劑成本,以含5%濃氨試液的甲醇溶液25 mL為提取溶劑、超聲處理30 min為最優方案。
3.1.3 色譜檢測條件篩選 分別考察水、甲醇、乙腈、甲(乙)酸等不同比例的流動相體系的分離效果。結果表明,以甲醇為有機相時,基線噪音較大,響應值偏低,且分離度不符合要求,采用乙腈為有機相可顯著改善上述問題。為改善待測組分色譜峰的峰形,嘗試在流動相中加入一定量的酸,當流動相為含0.1%甲酸的乙腈-0.1%甲酸水溶液時,目標組分定性、定量離子對峰型良好,與其他組分分離效率高。最終確定以含 0.1%甲酸的乙腈-0.1%甲酸溶液為流動相,并對其進行優化,建立梯度洗脫程序。
考察了不同柱溫(30、35、40 ℃)對供試品溶液中特征離子對m/z 943.6→635.5峰面積及分離度的影響,結果表明隨柱溫升高,目標峰面積及分離度均結果滿意,故采用40 ℃柱溫進行測定。
3.1.4 質譜條件篩選 分別考察了電噴霧正、負離子模式下對照品溶液和供試品溶液中目標色譜峰檢出情況,結果表明,采用負離子模式可檢出Nepasaikosaponin K的分子離子峰[M-H]-943.6、氯離子加合峰[M + Cl]-979.7和羧基加合峰[M + HCOO]-989.6,其中分子離子峰[M-H]-943.6 豐度最高(見圖1);正離子模式未檢測出Nepasaikosaponin K色譜峰。
選取豐度最高的分子離子峰[M-H]-943.6為母離子,篩選目標子離子,結果表明,主要碎片離子為[M-H-Glu]-781.5、[M-H-Rha]-797.4、[M-H-Glu-Rha]-635.5,裂解途徑見圖3。分別檢測對照品和供試品溶液中m/z 943.6→635.5、m/z 943.6→781.5、m/z 943.6→797.4特征離子對色譜峰,結果表明,三對特征離子均能檢出,其中943.6→635.5峰面積最大,信噪比高,分離度好;故本研究選擇m/z 943.6→635.5、943.6→781.5、943.6→797.4為Nepasaikosaponin K的定性檢測離子對,m/z 943.6→635.5為定量檢測離子對。

圖3 Nepasaikosaponin K化學結構式及碎裂方式示意圖
3.2 Nepasaikosaponin K在柴胡屬飲片中分布情況分析 本研究對北柴胡、南柴胡、藏柴胡、錐葉柴胡、竹葉柴胡等5種市售常見柴胡屬飲片中Nepasaikosaponin K含量測定。結果如表4所示,Nepasaikosaponin K雖然在上述5種柴胡中均存在,但藏柴胡與其他4種柴胡之間含量具有顯著性差異;其中藏柴胡含量平均值為486.4 μg·g-1,而在北柴胡、南柴胡、錐葉柴胡、竹葉柴胡中的含量平均值均在20 μg·g-1以下;藏柴胡中Nepasaikosaponin K含量約為其他種柴胡的25~140倍。因此,Nepasaikosaponin K可作為區別藏柴胡和其他種柴胡的差異標志物。
3.3 柴胡屬飲片藏柴胡摻偽比例測定方法探討 結果表明,藏柴胡摻偽比例在0%~100%范圍內的混合樣品中,Nepasaikosaponin K定量離子對m/z 943.6→635.5色譜峰峰面積與摻偽比例之間的線性關系良好。同時,本研究建立了藏柴胡摻偽比例計算公式,結果表明,不同柴胡屬模擬摻偽樣品的計算摻偽量與實際摻偽量的平均偏差在0.6%~1.4%之間,說明本研究建立的方法可實現對藏柴胡摻偽量的定量檢測,同時也為摻偽分析提供了新的思路。
3.4 市售柴胡屬飲片中藏柴胡摻偽情況分析及擬定限度研究 因藏柴胡性狀與北柴胡較為接近,且北柴胡市場需求量較大,目前藏柴胡多摻入或混充北柴胡使用。本次采集的疑似摻偽柴胡屬樣品均為北柴胡中摻雜部分藏柴胡,經測定,市售3批疑似摻偽樣品摻偽比例為14%~27%。
結合市場調研情況,其他種柴胡飲片中混有低于5%的藏柴胡可認定為非故意摻偽;且多種柴胡屬飲片中均含有微量的Nepasaikosaponin K,質譜可檢出,故本次研究以摻入5%藏柴胡的北柴胡模擬樣品的含量限度作為擬定限度。
綜上所述,本研究建立了以Nepasaikosaponin K為指標的柴胡屬飲片中藏柴胡摻偽檢測方法,該方法采用HPLC-MS/MS技術,可對北柴胡、南柴胡、藏柴胡、錐葉柴胡、竹葉柴胡等5種市售常見柴胡屬飲片中Nepasaikosaponin K進行定量檢測,操作簡便、專屬性強、準確度高。同時,采用該檢測方法,建立了柴胡屬飲片中藏柴胡摻偽比例計算公式,并以Nepasaikosaponin K為指標,開展了藏柴胡摻偽限度研究。本研究可用于柴胡屬飲片質量評價及真偽篩查,為柴胡質量監管提供了有力的技術支撐,同時也為中藥摻偽檢測技術研究提供新的思路與方法。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
