南寧地區遺傳代謝性肝內膽汁淤積癥患兒的臨床特征及基因分析
2022-11-09 02:02:46龐智東顏云盈徐江燕闕宇晨
肝臟 2022年10期
關鍵詞:基因突變
龐智東 顏云盈 徐江燕 闕宇晨
嬰兒膽汁淤積癥(infantile cholestasis,ICH)是嬰兒期最常見的肝膽疾病,雖然目前國內尚缺乏嬰兒膽汁淤積癥流行病學數據,但是已成為中國兒童肝病的首位住院原因[1]。若嬰兒膽汁淤積癥持續不緩解,可加速肝細胞損傷,隨病程進展而發展為肝纖維化和肝硬化,不但嚴重影響患兒的生長發育,甚至危及患兒健康及生命[2]。嬰兒膽汁淤積癥的病因較多,并且部分肝內膽汁淤積癥臨床表現不典型,常規檢查無特異性,臨床診斷困難。近年來隨著基因檢測技術的發展和臨床應用,其在新生兒遺傳性疾病的檢測或診斷以及部分常見病的輔助診斷方面應用越來越廣泛。基因檢測的應用使越來越多原因不明的嬰兒膽汁淤積癥得以確診,具有重要的臨床價值[3]。但是,目前國內關于基因檢測診斷嬰兒膽汁淤積癥的研究報道有限。本文對南寧地區疑似遺傳代謝性肝內膽汁淤積癥患兒的臨床特征及遺傳基因進行分析,為該地區遺傳代謝性嬰兒膽汁淤積癥的診斷和治療提供參考。
資料與方法
一、研究對像
選取2016年1月至2020年12月在南寧市婦幼保健兒科接受治療的116例疑似遺傳代謝性肝內膽汁淤積癥患兒作為研究對象。納入標準:①患兒在南寧地區出生;②年齡≤1歲;③臨床表現符合美國兒科學會推薦的膽汁淤積癥診斷標準[1],兼具膽汁淤積和肝病兩部分臨床征象;④均接受代謝性肝病相關基因檢測;⑤排除膽道閉鎖等器質性病變;⑥家長知情同意。以下患兒不參加本研究:①年齡≥1歲;②未接受代謝性肝病相關基因檢測;③存在膽道閉鎖等器質性病變的患兒;④家長知情但是不同意參與本研究。本研究得到醫學倫理委員會批準。
二、研究方法
(一)臨床資料的收集 采用自制調查表收集每例膽汁淤積癥患兒的臨床資料,包括性別、發病年齡、就診年齡、體重、喂養方式、皮膚黃染時間、大便顏色、肝功能、腹部彩超(肝、脾、膽囊)。其中,肝功能包括血清丙氨酸轉氨酶(alanine aminotransferase,ALT)、天冬氨酸轉氨酶(aspartate aminotransferase,AST)、γ-谷氨酰轉移酶(gamma-glutamyl transpeptidase,GGT)、堿性磷酸酶(alkaline phosphatase,ALP)、總膽汁酸(total bile acid,TBA)、總膽紅素(total bilirubin,TBil)、總蛋白(total protein,TP)、白蛋白(Albumin,Alb)、球蛋白(globulin,Glb)等指標。
(二)基因檢測 抽取患兒外周血,采用血液全基因組DNA 提取試劑盒(北京天根生化科技公司)按照說明書操作提取基因組DNA,制備全基因組文庫并進行質控。應用GenCap 液相捕獲目標基因技術,捕獲代謝性肝病相關基因的編碼外顯子區域,采用聚合酶鏈反應(PCR)對DNA進行擴增,采用Sanger進行驗證。
三、統計學處理
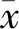
結 果
一、患兒的臨床特征
116例患兒中男67例(57.76%),女49例(42.24%),發病年齡為(1.0±0.5)個月;就診年齡為(3.4±2.7);體質量為(6.14±1.57)kg。母乳喂養例55例(47.41%),人工喂養26例(22.41%)和混合喂養35例(30.17%);皮膚染黃時間1~30 d,其中<3 d 47例(40.52%)、3~15 d 34例(29.31%),≥15 d 35例(30.17%);116例患兒全部為黃疸;35例(30.17%)患兒出現白陶土色便。ALT(214.40±72.36)U/L,AST(341.25±95.49)U/L,γ-GGT(116.03±57.36)U/L,ALP(546.06±245.94)U/L,TBA(141.52±72.36)μmol/L,TBil(154.36±64.42)μmol/L,TP(54.35±7.64)g/L,Alb(37.02±5.82)g/L,Glb(16.59±4.21)g/L。肝臟彩超顯示35例(30.17%)肝臟基本正常,81例(69.83%)肝臟腫大,腫大(1.65±0.84)cm;脾臟彩超顯示76例(65.52%)脾臟基本正常,40例(34.48%)脾臟腫大,腫大(0.57±0.51)cm;膽囊彩超顯示41例(35.34%)患兒膽囊基本正常,75例(64.66%)膽囊異常(發育異常或收縮欠佳)。
二、基因突變分析
有35例發生基因突變,其中SLC25A13 基因突變11例(31.43%),JAG1 基因突變8例(22.86%),ABCB11 基因突變7例(20.00%),HSD3B7 基因突變5例(14.29%),CFTR 基因和AKR1D1 基因突變各2例(5.71%)。基因突變患兒中有希特林蛋白缺乏癥11例(31.43%),Alagille綜合征8例(22.86%),進行性家族性肝內膽汁淤積癥2型7例(20.00%),先天性膽汁酸合成障礙1型4例(11.43%)先天性膽汁酸合成障礙2型3例(8.57%),囊性纖維化2例(5.71%)。見表1。
三、基因突變和非基因突變患兒臨床資料比較
基因突變膽汁淤積癥患兒ALT水平較非基因突變膽汁淤積癥患兒顯著升高(P<0.05),而兩組患兒的性別、發病年齡、就診年齡、體質量、喂養方式、皮膚染黃時間、白陶土色便、AST、γ-GGT、ALP、TBA、TBil、TP、Alb、Glb、肝臟彩超情況、脾臟彩超情況和膽囊彩超情況差異均無統計學意義(P>0.05)。見表2。

表1 膽汁淤積癥患兒基因突變情況

表2 基因突變和非基因突變患兒臨床資料的比較
討 論
嬰兒膽汁淤積癥是指由各種原因導致毛細膽管膽汁形成、分泌和(或)排泄異常,在肝細胞和膽管內膽汁淤積引起的肝臟疾病。嬰兒膽汁淤積癥兼具膽汁淤積和肝病兩部分臨床征象,膽汁淤積以膽紅素升高,皮膚、鞏膜黃染為主要表現,肝病以血清ALT和(或)AST水平增高等肝功能異常為主要表現。嬰兒膽汁淤積癥起始于膽汁酸淤積,進一步導致肝細胞增生和壞死,繼而發生膽管性肝纖維化和肝硬化,或肝功能衰竭而進入肝病終末期[4],是嬰兒期致死或致殘的重要原因之一。嬰兒膽汁淤積癥包括肝外膽汁淤積和肝內膽汁淤積。肝內膽汁淤積癥的病因復雜,有將近1/3的病因不明確,從而導致臨床預后差異顯著,早期診斷、早期治療可改善患兒預后[5-7]。高通量測序技術因其速度快、通量高等特點廣泛應用于臨床,在分子生物學研究及疾病診斷方面發揮巨大作用。考慮到肝內膽汁淤積癥是一種發病率較高的常染色體隱性遺傳代謝病,因此高通量測序技術對肝內膽汁淤積癥的診斷意義重大。本研究116例患兒,通過目標基因捕獲結合高通量測序技術證實有35例患兒發生基因突變,包括SLC25A13 基因突變、JAG1 基因突變、ABCB11 基因突變、HSD3B7 基因突變、CFTR 基因和AKR1D1 基因突變。
希特林蛋白缺乏癥是由 SLC25A13 基因突變引起的常染色體隱性遺傳病,該基因突變可導致位于肝細胞線粒體內膜的Citrin 蛋白即天冬氨酸/谷氨酸載體2型蛋白功能缺陷,從而導致各種代謝紊亂[8]。在本研究中有11例為希特林蛋白缺乏癥患兒,均測出有SLC25A13 基因突變。Takahashi等[9]研究表明,約70%的SLC25A13突變攜帶者為851 de14 突變,而約1/17嬰兒膽汁淤積性黃疸患兒攜帶851 de14突變。羅玲英等[10]通過檢測40例希特林蛋白缺乏癥導致的嬰兒肝內膽汁淤積癥患兒發現,SLC25A13突變基因主要包括851de14、IVS16ins3kb和IVS6+5GD>A等。白欣立等[11]對希特林蛋白缺乏癥導致新生兒肝內膽汁淤積癥的4例患兒的SLC25A13基因突變進行檢測,結果顯示突變包括c.851_854delGTAT、c.851_854delGTAT/c.115G>T、c.1064G>A/c.1157G>T和c.1078C>T/c.IVS4+6A>G。在本研究中,檢測到希特林蛋白缺乏癥導致新生兒肝內膽汁淤積癥的SLC25A13基因突變包括c.1078C>T/g.IVS4+6A>G、c.852_855del/c.615+5G>A、c.736delG/IVS16ins3kb、c.1095delT/c.1064G>A、c.G1064G>A/c.G1157G>T和g.IVS11+1G>A/g.IVS16ins3kb。因此SLC25A13是希特林蛋白缺乏癥導致的嬰兒肝內膽汁淤積癥的常見突變基因,但是各個研究檢測到的突變位點差異較大。
Alagille綜合征為常染色體顯性遺傳病,與JAG1 及 NOTCH2 基因變異有關[12],可累及肝臟、心臟、骨骼、眼和面部等多器官,典型肝臟病理表現為小葉間膽管缺乏,但是其臨床表現也不完全一致。本研究中有8例Alagille綜合征患兒,均為JAG1 基因突變,突變位點包括c.439+1G>A、c.3521C>T、c.1655C>T和c.2906T>C。王美娟等[13]對3例Alagille綜合征患兒基因突變檢測結果顯示也均為JAG1基因突變,突變位點與本研究結果一致。程映等[14]采用DNA 直接測序發現,Alagille綜合征患兒的JAG1基因存在c.2419delG突變,但是該突變位點在本研究中未發現。
進行性家族性肝內膽汁淤積癥為嬰兒期或兒童期起病的常染色體隱性遺傳性疾病,根據致病基因不同分為1型(ATP8B1基因突變導致家族性肝內膽汁淤積相關蛋白1缺陷)、2型(ABCB11基因突變導致膽鹽排泄泵缺陷)和3型(ABCB4基因突變導致多耐藥糖蛋白3缺陷)。本研究篩選出7例ABCB11 基因突變導致的進行性家族性肝內膽汁淤積癥2型,基因突變的位點有c.2594C>T/c.239T>C、c.3334G>T/c.908+1G>A和c.2594C>T/c.667C>T,與王美娟等[13]報道的ABCB11 基因突變位點一致。金萌等[15]報道了3例ABCB11 基因突變導致的進行性家族性肝內膽汁淤積癥2型患兒,基因突變的位點包括c.2594C>T/c.239T>C、c.2332G>A/c.2077C>G、c.1493T>C/c.1331T>C,與本研究僅有c.2594C>T/c.239T>C突變位點一致。
先天性膽汁酸合成障礙是一類由于合成膽汁酸過程中的酶缺陷引起,大多屬于常染色體隱性遺傳病,也可以是自身基因自發突變導致,臨床罕見。本研究篩選出4例先天性膽汁酸合成障礙1型,發生HSD3B7 基因突變,突變位點為c.557C>T/c.968C>G和c.1031 A>G;3例先天性膽汁酸合成障礙2型患兒,CFTR 基因和HSD3B7 基因發出突變,HSD3B7 基因突變位點為c.396C>A/c.722A>T,CFTR 基因突變位點為c.317_324del/c.1456G>T;2例囊性纖維化患兒,發生AKR1D1 基因突變,突變位點為c.158A>G/c.658A>C和c.579+2delT/c.853C>T。
通過分析基因突變和非基因突變患兒的臨床資料結果顯示,除基因突變患兒血清ALT水平稍高于非基因突變患兒外,其他臨床資料包括性別、發病年齡、就診年齡、體重、喂養方式、皮膚染黃時間、白陶土色便、AST、γ-GGT、ALP、TBA、TBil、TP、Alb、GLB、肝臟彩超情況、脾臟彩超情況和膽囊彩超情況差異均無統計學意義,因此基因突變和非基因突變患兒的基本臨床征象基本相似,通過臨床征象診斷基因突變的遺傳代謝性肝內膽汁淤積癥難度比較大。
因此,南寧地區發生基因突變的肝內膽汁淤積癥患兒病因繁多,臨床特征無顯著特征,通過高通量測序技術對臨床疑似遺傳代謝病的肝內膽汁淤積癥患兒的診斷有重要價值。
利益沖突聲明:所有作者均聲明不存在利益沖突。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
