夏蠣金消顆粒的質量控制方法研究*
2022-10-27 00:30:22河北中醫學院第一附屬醫院腫瘤二科
河北中醫藥學報 2022年5期
河北中醫學院第一附屬醫院腫瘤二科
范煥芳 閆嬌嬌△ 李國川△△ 馬 盼 李德輝 韓長輝 王崢嶸 王 驍 魏莉瑛(石家莊 050011)
提要 目的:建立夏蠣金消顆粒的質量控制方法,為該制劑的質量控制標準提供依據。方法:采用薄層色譜法(TLC)對顆粒處方中陳皮、黃芪、石見穿、當歸、紫菀、甘草進行鑒別;高效液相色譜法(HPLC)對橙皮苷進行含量測定、方法學驗證。結果:TLC法分離度高,展開效果好,陰性無干擾;橙皮苷對照品含量在1.015~5.075 μg之間時與峰面積線性關系良好,精密度、穩定性、重復性試驗RSD值均小于2.0%,加樣回收試驗RSD值為0.77%,平均回收率為98.17%。3批夏蠣金消顆粒中橙皮苷含量分別為1.37、1.38、1.48,平均RSD值為1.28%。結論:該方法準確、穩定,重現性高,可為夏蠣金消顆粒的質量標準提供實驗依據。
夏蠣金消顆粒是河北省中醫院腫瘤二科治療肺癌的中藥復方制劑,由清半夏、陳皮、茯苓、石見穿、牡蠣、黃芪、沙參、當歸、蜜紫菀、蜜款冬花、貓爪草、川貝母、生甘草13味中藥組成,具有化痰解毒、益氣養陰的功效,長期臨床研究表明,夏蠣金消顆粒能明顯改善肺癌患者咳嗽、咳痰、胸痛等癥狀,并且降低化療后毒副反應[1]。夏蠣金消顆粒在臨床應用廣泛、療效確切,但目前缺乏標準的質量評價,為了進一步明確其中的有效成分、含量,建立全面可靠的質量控制方法,本研究除利用薄層色譜法(TLC)對顆粒處方中陳皮、黃芪、石見穿、當歸、紫菀、甘草進行定性鑒別外,還采用高效液相色譜法(HPLC)對顆粒中含量豐富、專屬性強的指標性成分橙皮苷進行定量分析,為夏蠣金消顆粒的質量標準提供實驗依據。
1 材料
1.1 儀器 KQ2200E型超聲清洗器(昆山市超聲儀器有限公司),ZF-90型暗箱式紫外透射儀(上海顧村電光儀器廠),電熱鼓風干燥箱(上海一恒科學儀器有限公司),DK-98-II型電熱恒溫水浴鍋(天津市泰斯特儀器有限公司),FA2104N型電子天平(上海民橋精密科學儀器有限公司),LC-15C島津液相色譜儀(蘇制05000111號)。
1.2 藥品與試劑 橙皮苷(批號110721-201014,20 mg)、黃芪甲苷(批號110781-201717,20 mg)來自中國藥品生物制品檢定所。對照藥材:當歸(批號120927-201617,0.5 g)、紫菀(批號120956-200504,2 g)、甘草(批號120904-201620,1 g)、黃芪(批號120974-201612,1 g)、石見穿(批號121579-201001,10 g),均來自中國食品藥品檢定研究院。夏蠣金消顆粒樣品(批號:20190305、20190315、20190326)及陰性對照所用藥材均來自河北省中醫院。乙腈、磷酸均為色譜純,水為蒸餾水,乙酸乙酯、正丁醇、甲醇、甲酸、三氯甲烷、甲苯、正己烷等試劑均為分析純。
1.3 實驗用薄層板 (1)硅膠G板(青島海洋化工廠);(2)自制含羧甲基纖維素鈉(CMC)的薄層板:取5 g CMC,緩慢加入盛有1 000 mL蒸餾水的三角瓶中,置于磁力攪拌器上,溫度70 ℃~80 ℃,轉速適中,溶解后靜置。按硅膠G∶上述溶液=1∶3的比例,用自動薄層制板器制成厚度4 mm,20×10 cm的薄層板,晾干,于105 ℃烘干,放置干燥箱中;(3)自制含1%氫氧化鈉的薄層板:取上述(2)中制備好的溶液100 mL,加入1%氫氧化鈉,溶解后,其余方式同(2)。
2 薄層鑒別方法及結果
2.1 夏蠣金消顆粒的制備 夏蠣金消顆粒采用的是傳統濕法制粒,生藥加8倍水,煎煮2次,每次2 h,藥液濃縮后經60%乙醇沉淀、噴霧干燥,加輔料糊精、潤濕、過篩即得[2]。
2.2 薄層鑒別
2.2.1 陳皮的薄層色譜鑒別[3]:取3批次顆粒細粉各5 g,加甲醇50 mL,充分震蕩使其混勻,放置水浴上加熱回流30 min,濾過,濾液蒸干后,加入5 mL甲醇溶解殘渣,制備供試品溶液;取不含陳皮的陰性藥材,同法制成陰性對照溶液;另稱取0.407 5 g橙皮苷對照品,加1 mL甲醇,制成濃度為0.407 5 mg/mL的對照品溶液。吸取供試品溶液2 μL、陰性對照溶液2 μL、對照品溶液3 μL,在硅膠G薄層板上點樣,展開劑為乙酸乙酯-甲醇-水(9∶1.5∶1),展距約7 cm,晾干后噴三氧化鋁顯色劑,放置于紫外線(365 nm)下檢視,對照品色譜、供試品色譜在相同位置上,顯示相同顏色的斑點,陰性無此斑點,見圖1。

注:1~3.三批次供試品;4.陰性對照;5.橙皮苷對照圖1 陳皮薄層色譜圖
2.2.2 黃芪的薄層色譜鑒別[4]:取3批次顆粒細粉各5 g,加入30 mL 50%甲醇,30 min超聲處理,紗布濾過,濾液蒸干,以50 mL水溶解殘渣后,移入分液漏斗中,用水飽和的正丁醇溶液50 mL萃取2次,合并萃取液,用50 mL氨試液洗滌2次,取正丁醇液,濾過,濾液蒸干,殘渣加2 mL甲醇溶解,作供試品溶液;取不含黃芪的陰性藥材、黃芪對照藥材5 g,參照供試品溶液配制方法制成陰性對照溶液、對照藥材溶液。另取黃芪甲苷2 mg,加甲醇2 mL,制成濃度1 mg/mL的對照品溶液。吸取供試品溶液5 μL、陰性對照溶液2 μL、對照藥材溶液3 μL、黃芪甲苷對照品溶液10 μL,在自制的以羧甲基纖維素鈉(CMC)為黏合劑的硅膠G薄層板上點樣,展開劑為氯仿∶甲醇∶水(14∶5∶1)的下層溶液,展距約9 cm,顯色劑為10%硫酸乙醇,105 ℃加熱至斑點顯色清晰,后置于紫外線(365 nm)下檢視,供試品色譜在與黃芪對照藥材、黃芪甲苷對照品色譜相同位置上,顯示顏色相同的主斑點,陰性無此斑點,見圖2。

注:1~3.三批次供試品;4.陰性對照;5.黃芪對照藥材;6黃芪甲苷圖2 黃芪薄層色譜圖
2.2.3 石見穿的薄層色譜鑒別[5]:取3批次顆粒細粉各5 g,加30 mL 75%甲醇,置水浴加熱回流1 h,濾過,蒸干濾液,殘渣加入3 mL 75%甲醇溶解,作供試品溶液;取不含石見穿的陰性藥材、石見穿對照藥材5 g,按照上述供試品溶液制備方法制成陰性對照品溶液、對照品溶液。分別吸取上述溶液2 μL,在硅膠G薄層板上點樣,展開劑為乙酸乙酯-三氯甲烷-甲苯-甲醇-甲酸(5∶3∶2∶1∶2),展距約10 cm,顯色劑為三氯化鐵-鐵氰化鉀試液,于日光下進行檢視,供試品色譜在與石見穿對照藥材色譜相應位置上,顯示顏色相同的主斑點,陰性無此斑點,見圖3。
2.2.4 當歸的薄層色譜鑒別[6]:取3批次顆粒細粉各5 g,加入30 mL正己烷,30 min超聲處理,濾過,將濾液濃縮到2 mL,作供試品溶液;取不包含當歸的陰性藥材、當歸對照藥材0.25 g,參照供試品溶液制備方法制備陰性對照溶液、對照品溶液。吸取供試品溶液30 μL、陰性對照溶液2 μL、對照品溶液2 μL,在硅膠G薄層板上點樣,展開劑為正己烷-乙酸乙酯(8∶1),展距約7 cm,放置于紫外線(365 nm)下檢視,供試品色譜在與當歸對照藥材色譜相應位置上,顯示顏色相同的斑點,陰性無此斑點,見圖4。
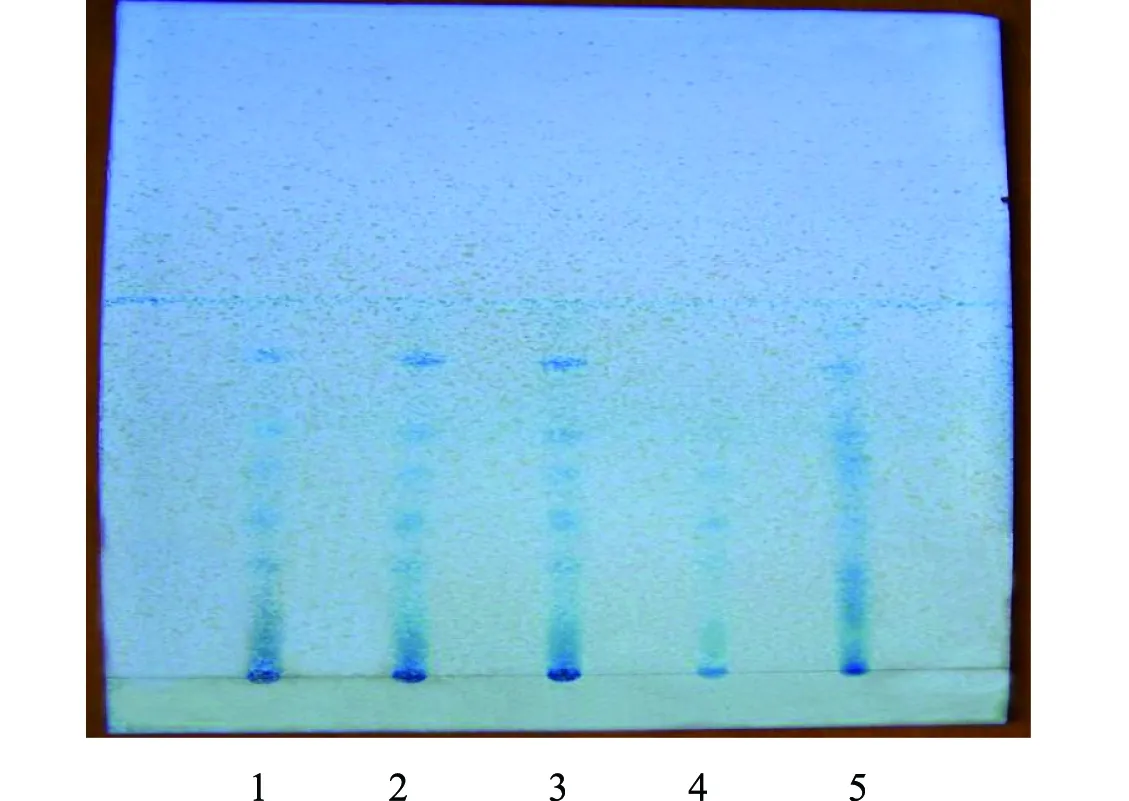
注:1~3.三批次供試品;4.陰性對照;5.石見穿對照藥材圖3 石見穿薄層色譜圖
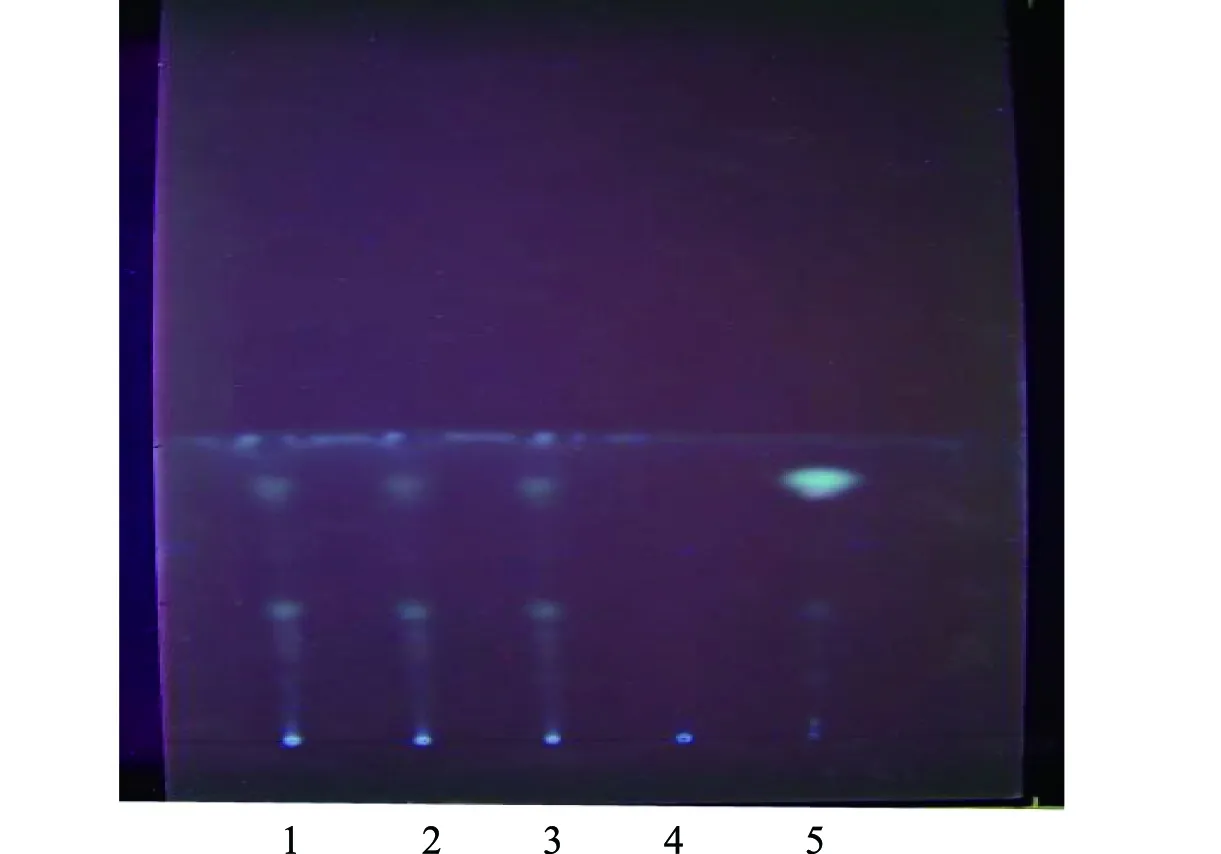
注:1~3.三批次供試品;4.陰性對照;5.當歸對照藥圖4 當歸薄層色譜圖
2.2.5 紫菀的薄層色譜鑒別[7]:取3批次顆粒細粉各5 g,加25 mL甲醇,超聲處理30 min,濾過、濾液蒸干,殘渣加入1 mL乙酸乙酯溶解,作供試品溶液;取不含紫菀的陰性藥材、紫菀對照藥材0.25 g,按供試品溶液制備方法,制成陰性對照溶液、對照品溶液。分別吸取供試品溶液15 μL、陰性對照溶液15 μL、對照品溶液5 μL,在自制的以CMC為黏合劑的硅膠G薄層板上點樣,展距約9 cm,噴10%硫酸乙醇顯色劑,105 ℃加熱至斑點清晰,放置于紫外線(365 nm)下檢視,供試品色譜在與紫菀對照藥材色譜相應位置上,顯示顏色相同的主斑點,陰性無此斑點,見圖5。
2.2.6 甘草的薄層色譜鑒別[8]:取3批次顆粒細粉各5 g,加入25 mL乙醚,置水浴上加熱回流30 min,濾過,濾液蒸干,殘渣加入5 mL甲醇溶解,作供試品溶液;取不含甘草的陰性、甘草對照藥材0.5 g,按供試品溶液制備方法制成陰性對照溶液、對照品溶液。吸取供試品溶液30 μL、陰性對照溶液8 μL、對照品溶液5 μL,在自制的含1%氫氧化鈉的硅膠G薄層板上點樣,展開劑為醋酸乙酯-冰醋酸-甲酸-水(15∶1∶1∶2),充分飽和15 min,展距約8 cm,以10%硫酸乙醇為顯色劑,105 ℃加熱至斑點清晰,放置于紫外線(365 nm)下檢視,供試品色譜在與甘草對照藥材色譜相應位置上,顯示顏色相同的斑點,陰性無此斑點,見圖6。
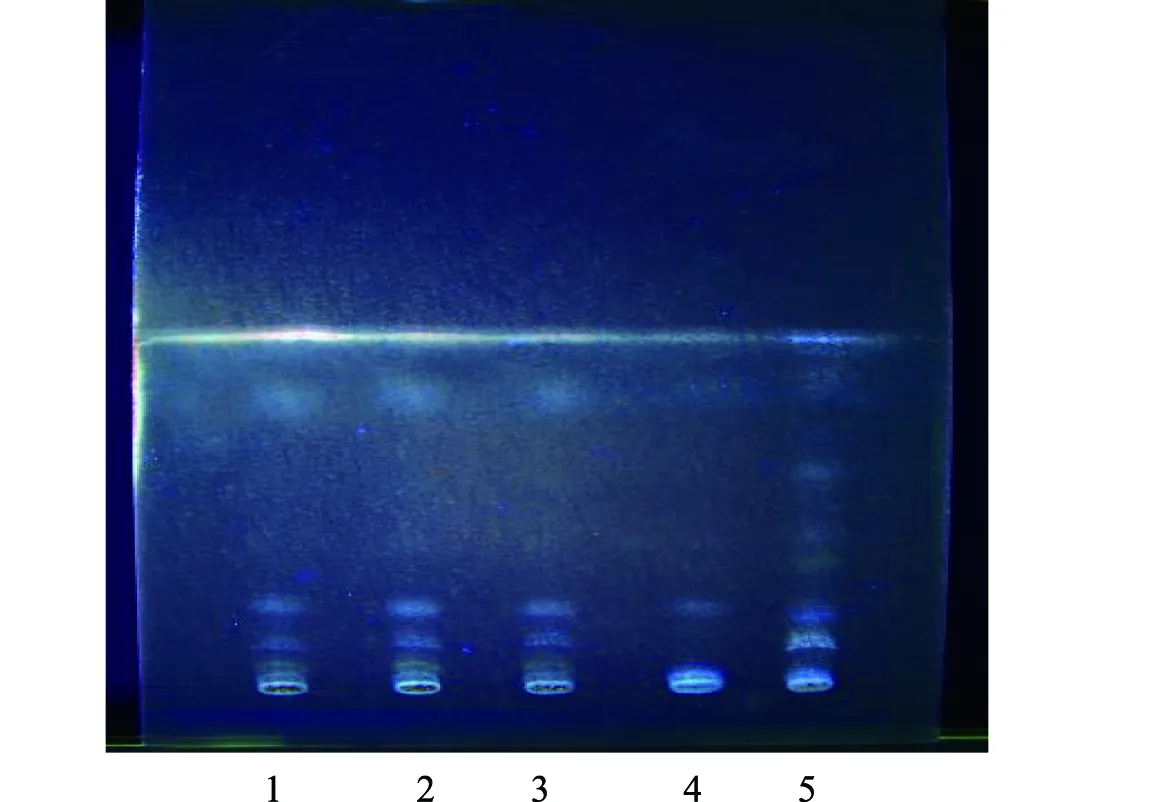
注:1~3.三批次供試品;4.陰性對照;5.紫菀對照藥材圖5 紫菀薄層色譜圖
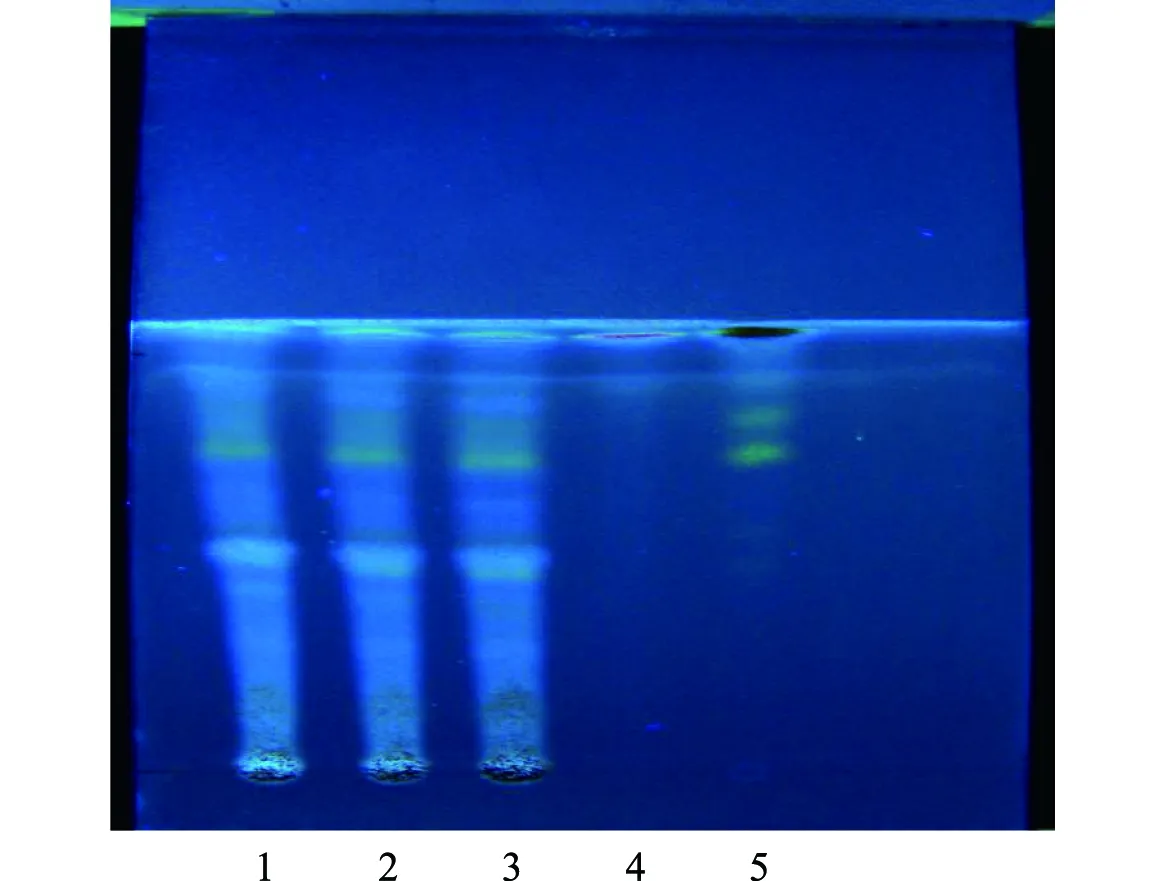
注:1~3.三批次供試品;4.陰性對照;5.甘草對照藥材圖6 甘草薄層色譜圖
3 含量測定方法及結果
3.1 色譜條件 色譜柱:C18柱(5 μm,4.6×200 mm);流動相:乙腈-0.1%磷酸(20%∶80%);檢測波長為283 nm;理論板數按橙皮苷峰計算應不低于2 000;流速:1.0 mL/min;柱溫:25 ℃;進樣量:10 μL。
3.2 溶液制備 (1)對照品溶液:稱取適量橙皮苷對照品,放置于容量瓶中,加適量甲醇,超聲5 min,加甲醇定容至10 mL,過水濾膜,制成質量濃度為0.14 mg/mL的對照品溶液。(2)供試品溶液:取供試品顆粒,研磨后過80目篩,取細粉1 g,加水適量,超聲5 min,定容至25 mL,取上清液,過水濾膜,即得。(3)陰性對照溶液:取不含陳皮的陰性對照,陰性對照溶液制備方法參照上述供試品溶液。
3.3 專屬性考察 精密吸取對照品溶液、供試品溶液、陰性對照溶液各10 μL,注入高效液相色譜儀中,陰性對照色譜圖中顯示,在與對照品溶液、供試品溶液色譜圖的橙皮苷峰相應位置處無相同吸收峰,結果表明專屬性良好,結果見圖7。
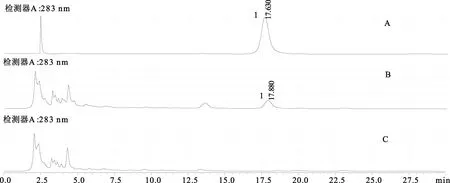
注:A.對照品溶液;B.供試品溶液;C.陰性對照溶液;1.橙皮苷圖7 HPLC圖
3.4 線性關系考察 取橙皮苷對照品適量,加甲醇溶解并定容至10 mL,配制濃度為203 μg/ mL的橙皮苷對照品溶液,按上述色譜條件進行測定,設定分別進樣5、10、15、20、25 μL,以對照品含量為橫坐標,峰面積為縱坐標,得回歸方程:y=336 353.62x-20 388.7(r=0.999 9) ,說明在對照品含量在1.015~5.075 μg之間,與峰面積線性關系良好,結果見表1、圖8。

表1 線性試驗結果
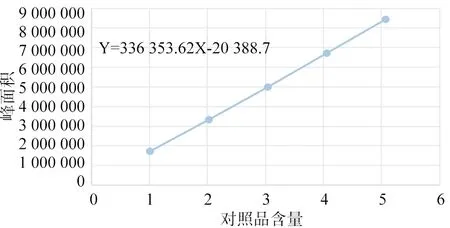
圖8 標準曲線圖
3.5 精密度試驗 取濃度為0.14 mg/mL的同一對照品溶液,進樣量為10 μL,重復測6次,計算峰面積,最終得到橙皮苷峰面積的RSD值為0.798%,結果表明,設備精密度良好。
3.6 穩定性考察 稱取同一批次顆粒細粉1 g,加水超聲處理,定容至25 mL,于0、2、4、8、12 h分別進樣10 μL,測定其峰面積,計算出RSD值為1.351 8%(n=5),結果表明樣品溶液在12 h內穩定。
3.7 重復性考察 稱取同一批次顆粒細粉1 g,加水超聲處理,定容至25 mL,平行6份,分別進樣10 μL,計算橙皮苷峰面積,得出RSD值為0.85%(n=6),結果表明該測定方法重復性良好。
3.8 加樣回收實驗 精密稱取同一批次顆粒細粉9份,每份約0.25 g,每份樣品加入適量橙皮苷對照品溶液,加水定容至5 mL,進行含量測定,計算加樣回收率,結果詳見表2,平均回收率為0.981 7%,RSD值0.77%(n=9),加樣回收率符合要求,該方法回收率良好。
3.9 3批樣品含量測定 取3批次樣品顆粒,按“3.2溶液制備”方法配制供試品溶液,對3批次供試品溶液中(批號:20190305、20190315、20190326)橙皮苷進行含量測定,結果見表3。從3批次橙皮苷含量可見,每克樣品顆粒中橙皮苷含量在1.37~1.48 mg,含量相對穩定,但考慮到制備工藝、藥品存放時間等因素的影響,按含量下降幅度20%計算,每克樣品中橙皮苷含量不得低于1.09 mg。
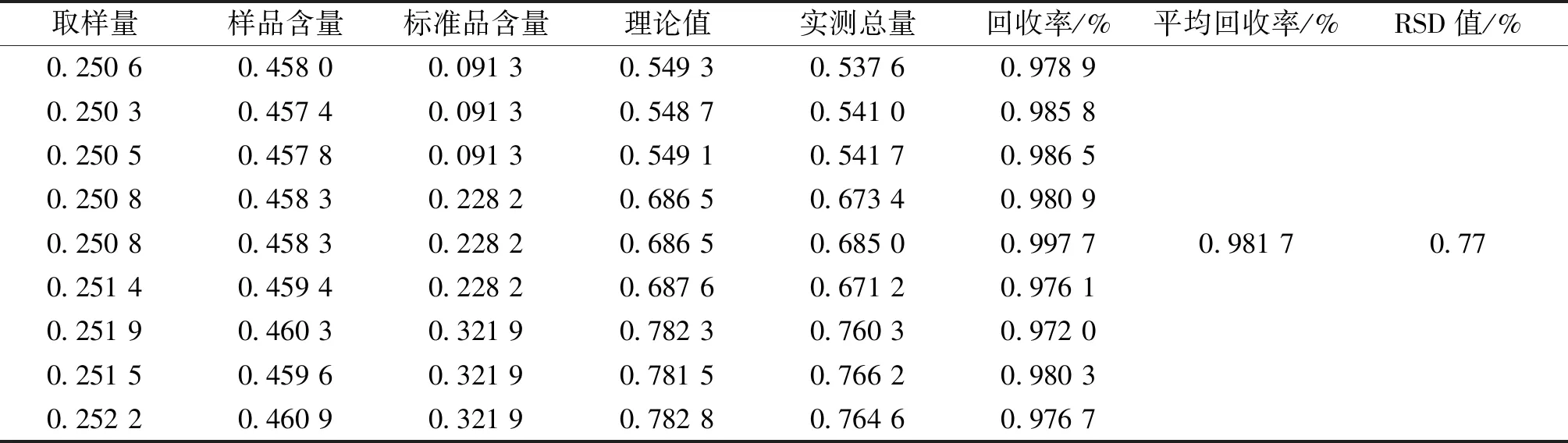
表2 加樣回收試驗結果 (mg)

表3 3批樣品含量測定 (mg/g)
4 討論
4.1 薄層色譜法(TLC) TLC在《中華人民共和國藥典》中應用普遍且逐漸完善,在中藥質量控制中直觀、顯著、分離速度快,占據重要位置[9]。《中華人民共和國藥典》2020年版中有陳皮、黃芪、當歸、紫菀、甘草的薄層鑒別方法,但因劑型、制備工藝及藥物相互作用的影響,操作步驟也不能單純套用,本方法在薄層板的選擇、點樣方式、展開劑選擇方面進行改良,最終得到了分離度高、重現性好的薄層色譜。
在黃芪、紫菀、甘草的薄層鑒別中,初期色譜均出現拖尾,展出效果差,結合薄層板硅膠性質、黏合劑、pH值的差異,最終選用自制含CMC的硅膠G板、1%氫氧化鈉的硅膠G板,結果斑點清晰。在黃芪、紫菀薄層鑒別中,點樣方式根據多次顯色結果進行調整,由圓點點樣改為橫線少量多次點樣,此方法分離度高,斑點清晰,無明顯拖尾[10]。當歸的薄層鑒別時,將極易揮發的乙醚改為用正己烷超聲提取,正己烷-乙酸乙酯為展開劑,在此條件下,斑點分離清晰,無陰性干擾。黃芪展開劑選擇氯仿∶甲醇∶水的下層溶液,展開劑比例13∶7∶2,存在斑點重疊現象,最終調整為14∶5∶1,最終發現在此條件下,斑點分離度好,無陰性干擾。
4.2 高效液相色譜法(HPLC) 陳皮、半夏之二陳湯,具有理氣健脾、燥濕化痰的功效,為治痰之代表方劑,也被廣泛作為基礎方化裁,應用于肺癌的基礎及臨床研究中[11-13]。陳皮中含有多種豐富的化學成分,如黃酮類、萜類與揮發油、生物堿類等,有減少炎癥損傷,改善微循環,保護肺組織細胞等作用[14]。陳皮中指標成分橙皮苷含量極高,其有抗癌、抗炎、誘導細胞凋亡的作用[15]。在前期制粒工藝實驗中,就以橙皮苷為考察指標,在保證出干膏率同時最大程度兼顧到夏蠣金消顆粒中的有效成分[2]。測定證實,陳皮的指標成分橙皮苷提取工藝簡便,專屬性高、含量穩定且無陰性成分的干擾,能有效確保顆粒中有效成分達標、質量穩定。
色譜條件選擇:在200~400 nm波長段對橙皮苷對照品、供試品溶液進行掃描,結合相關研究[16],橙皮苷檢測波長為283 nm時有最大吸收。流動相參考《中國藥典》及有關文獻研究,考察甲醇-水、乙腈-磷酸、乙腈-水等系統,最終發現流動相為乙腈-0.1%磷酸(20%∶80%)時,分離效果良好,峰形對稱[17]。
提取溶液選擇:HPLC進行橙皮苷的含量測定時,分別采用50%甲醇、水2種方式對樣品進行處理,對比發現水比50%甲醇溶解性好,兩者所測得積分面積差異不大,考慮到顆粒本為水溶性物質,又處于安全性的考慮,故采用水提取的方式,經驗證,本研究所建立方法,專屬性強,線性關系及重復性良好,精密度、穩定性良好。
綜上所述,本研究利用TLC、HPLC對夏蠣金消顆粒進行定性、定量分析,該方法可操作性強、重復性好、應用廣泛,可為夏蠣金消顆粒的標準質量控制提供實驗依據。
