整合的HBV DNA在慢性乙型肝炎患者病毒復制及慢性感染維持中的潛在作用
2022-10-15 04:13:28李玉坤顧智強姜倩倩陳香梅魯鳳民
肝臟 2022年9期
關鍵詞:血清
李玉坤 顧智強 姜倩倩 陳香梅 魯鳳民
慢性感染是乙型肝炎病毒(HBV)最主要的致病形式。而以微小染色體形式存在于感染肝細胞核的長度3.2 kb的共價閉合環狀DNA(cccDNA)對于病毒復制及慢性感染的持續至關重要,并與慢性乙型肝炎(CHB)難以治愈和抗病毒治療停藥后的病毒反彈及疾病復發相關。作為病毒復制的源頭,cccDNA可以轉錄出超基因組全長的3.5 kb前基因組RNA(pgRNA)和亞基因組RNA(2.4 kb、2.1 kb和0.7 kb RNAs)[1],其中pgRNA既是編碼病毒核心蛋白(core)和具有逆轉錄酶活性的病毒DNA聚合酶蛋白(polymerase, pol)的mRNA,也是在由core蛋白構成的核衣殼內pol蛋白逆轉錄酶作用下合成子代病毒基因組——松弛環狀DNA(rcDNA)負鏈的模板[2]。在逆轉錄過程中,pol蛋白需要完成三次正確的跳轉才能產生子代rcDNA,若pol蛋白未發生第二次跳轉,而在原位啟動正鏈DNA的合成,則會形成雙鏈線性DNA(Double strand linear DNA,dslDNA)[3]。本文回顧了現有的與HBV DNA整合相關的知識體系,特別是其能否通過產生和提供病毒包膜蛋白HBsAg及羧基端缺失的HBx蛋白等參與支持病毒的復制,及其在慢性HBV感染維持中的潛在作用。
一、HBV DNA整合的分子機制及表達特征
目前已知dslDNA是整合HBV DNA的前體和來源,可通過非同源末端連接或微同源末端連接等方式隨機地整合至宿主DNA雙鏈斷裂處[4]。這里,我們通過對本實驗室前期數據及網上可用的HBV整合的病毒斷點數據的再分析,對整合的病毒片段斷點的分布特征提出了新的觀點。不同于既往的整合病毒DNA斷點分布于DR1及DR2間的籠統敘述,我們認為正確的表述應該是:下游斷點多集中在dslDNA下游端點的DR1殘存(1827nt,不同基因型間的脫氧核苷酸序號可能存在差異,下同)周圍,上游斷點則位于pgRNA 5'端殘留起始(1818nt)至上游DR1(含該DR1的1824-1834nt重復序列)區域附近(圖1)。這一表述與dslDNA的兩端斷點一致,更符合dslDNA為整合病毒DNA前體(precursor)的推測[5-6]。而之所以在實測的臨床樣本中出現斷點自dslDNA兩端內移的現象,多是因為整合的病毒-宿主融合位點微小區域的染色體不穩定性[2],及伴隨著攜帶整合片段肝細胞在肝臟炎癥壞死后的代償性克隆擴增帶來的病毒序列的微小缺失等改變所致。
從眾多已知的整合病毒DNA的兩端斷點推測,整合的HBV DNA序列多保留有完整的編碼大、中、小表面抗原的preS/S區及羧基端部分缺失的X基因區(圖1),該推測被近期的一系列基因組長測序結果所證實[7]。由此以來,整合的HBV DNA能夠在其自身啟動子的作用下轉錄出對應的2.4、2.1 kb 轉錄本及能夠翻譯出羧基端缺失并融合了部分宿主基因組序列的X基因轉錄本。既往研究也證實,整合HBV DNA可以支持乙型肝炎表面抗原(Hepatitis B surface antigen,HBsAg)和羧基端截短的HBx蛋白的表達[8-9]。同時,由于啟動子移位無法轉錄出超基因組全長的3.5 kb的pgRNA,“有缺陷”的整合HBV DNA不能單獨支持子代病毒基因組rcDNA的產生[10]。
二、HBV DNA整合與肝細胞癌發生
由于病毒DNA整合入宿主基因組事件可發生于包括感染早期在內的任何階段,且其在人染色體上整合具有隨機性,不同自然病程的CHB患者肝組織中均可以檢測到獨特的病毒-宿主DNA融合的整合事件[11]。目前關于HBV整合的研究多聚焦于整合事件的致肝細胞癌(HCC)作用。機理上,已知HBV DNA整合可通過以下三種主要機制促癌:(1)擾亂宿主癌基因的表達及其功能,包括細胞增殖等促癌基因如TERT、CCNE1和MLL4等的激活[12],及抑癌基因的功能失活等[5];(2)促進宿主染色體特別是整合位點局部基因組的不穩定性[13-14];(3)野生型和截短/融合病毒蛋白(HBx、preS/S)的致癌作用[15]。一般認為,HBV DNA整合的上述致HCC作用是與病毒所致肝臟的炎癥微環境相互作用的綜合結果,炎癥所致肝細胞的死亡誘導了殘余肝細胞的再生,而攜帶上述整合的肝細胞會因具有增殖優勢而被克隆性擴增,并積累更多的突變最終導致HCC的發生。
三、HBV DNA整合對病毒復制的影響及其慢性感染維持中的可能作用
目前,人們對于整合的HBV在病毒復制及慢性HBV感染維持中的作用仍然知之甚少。誠如前面所提,由于整合的HBV DNA前體dslDNA的全長僅為3.2 kb,無法轉錄出3.5 kb的pgRNA以支撐病毒DNA復制,故HBV DNA的整合事件被認為是錯誤跳轉帶來的副產品,這很容易讓人認為整合的HBV DNA并不參與病毒的復制。但對此也有人持不同的觀點。近期Erken等[16]建立了檢測CHB患者肝組織中整合HBV DNA水平的半定量巢氏Alu-PCR技術,并通過整合HBV DNA水平與患者血清病毒學指標的相關性分析,以期找出HBV DNA整合參與病毒復制的潛在證據。盡管我們的工作提示Erken等[17]采用的檢測整合HBV轉錄本的RT-Alu-PCR技術可能會漏掉大部分整合HBV的轉錄本,但仍應重視HBV DNA整合在病毒復制和慢性感染維持中的作用[18]。據此,本文總結了既往的研究發現,并結合我們自己的工作,嘗試從以下四方面進一步闡述HBV整合在病毒復制及慢性感染維持中的潛在作用。
(一)整合來源的HBsAg可為病毒復制提供包膜并維持持續感染 慢性HBV感染者血清HBsAg有cccDNA及整合的HBV DNA兩個來源,特別是對于乙型肝炎e抗原(HBeAg)陰性患者,整合的HBV DNA可能是其血清HBsAg的主要來源[19]。不僅如此,Freitas等發現,存在HBV DNA整合的PLC/PRF/5細胞系可以為丁型肝炎病毒(HDV)提供功能性包膜蛋白[20]。有關整合來源的HBsAg支持病毒包膜形成的實驗證據還包括:由于pol蛋白的逆轉錄酶活性缺乏矯正閱讀功能,HBV基因組(cccDNA)的preS/S區可有高頻的可導致HBV分泌和感染缺陷的突變發生[21-22],但在這些CHB患者體內卻可檢測到preS/S區突變毒株的持續存在[23],提示整合HBV DNA來源的功能性HBsAg可能彌補了preS/S區突變毒株的缺陷以維持該類毒株的持續感染。如果存在于肝細胞核內的cccDNA分子在不同的時空的某一節點僅轉錄一種病毒轉錄本,我們或許可以想象隨著疾病進展和病毒與宿主免疫的博弈,HBeAg陰性患者部分肝細胞核內殘留的數量顯著減少的cccDNA可能只具備轉錄pgRNA的能力了。此時,來自整合HBV DNA的病毒包膜蛋白將協助其形成具有感染能力的子代病毒顆粒。當然,該推測尚需更多的實驗證據的支持。
(二)整合的HBV DNA促進HBV復制的可能機制 整合HBV DNA表達的野生型或突變型HBsAg均可引起內質網應激[24]。Wang等[25]研究發現,HBV可利用內質網應激所啟動的自噬作用來增強HBsAg的產生及HBV的復制和分泌。此外,HBx通過促進宿主SMC5/SMC6復合物的降解,增強cccDNA的轉錄和病毒復制[26],為新合成cccDNA獲得轉錄活性和活性維持及病毒的持續復制所必須[27]。此外,作為共轉錄調控因子,HBx的羧基端(51-154aa)含有可轉錄激活cccDNA的反式激活結構域[28]。需要注意的是,整合過程中dslDNA的末端常發生缺失突變,由dslDNA兩端向內缺失最長可達200 bp[29],對應產生羧基端不同程度截短的CtHBx,其對于cccDNA的轉錄調控作用可能會不盡相同,有待進一步研究。
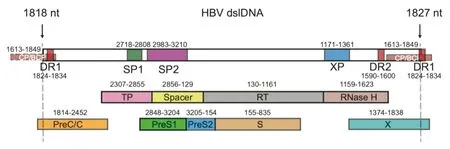
圖1 經典雙鏈線性DNA的序列特征(以HBV C基因型為例)
(三)整合HBV DNA為HBeAg陰性患者HBsAg主要來源的可能機制 盡管高水平的HBeAg和HBsAg具有免疫抑制作用[30-31],但HBeAg的免疫原性可能更強。因此,在慢性HBV感染的自然進程中,往往會發生HBeAg的陰轉,提示宿主免疫應答更傾向于清除表達HBeAg的cccDNA轉錄活躍的肝細胞,這一過程往往會誘發殘存肝細胞的代償性增殖,一方面會導致cccDNA池的進一步丟失,一些存在整合的肝細胞則可能會因代償性增殖并進一步克隆性擴增而逐漸增加[32-33]。在長期的疾病進展過程中,患者的HBeAg發生陰轉或血清學轉換,整合的HBV DNA逐漸成為其HBsAg的主要來源[34-35]。
(四)整合HBV DNA與CHB患者難以達到臨床治愈有關 無論是自發,還是經過抗病毒治療,血清HBsAg消失的CHB患者往往有更好的近期和遠期結局,并被定義為臨床治愈[36]。盡管人們知道血清HBsAg有cccDNA和整合HBV DNA兩個來源,但靶向HBV轉錄本3'端共同序列的首個RNA干擾候選藥物ACR-520在HBeAg陰性的慢性HBV感染黑猩猩中失去顯著抑制血清HBsAg的作用[37]。究其原因在于ACR-520靶向的mRNA序列位于病毒整合下游斷點之后,對整合來源pres/s轉錄本并無干擾作用[37]。最近高志良團隊對CHB患者的HBV DNA整合分析結果證實,與未實現臨床治愈者相比,臨床治愈患者肝組織內整合的HBV DNA有著極為明顯的減少[38],也說明了整合HBV DNA來源的HBsAg的存在使患者難以達到臨床治愈。
四、小結
HBV DNA整合一直被認為是HCC發生的主要危險因素之一,但隨著對HBV DNA整合的分子機制和致病機理的進一步認識,確證了盡管HBV DNA整合不會產生具有復制能力的轉錄本,但可以表達大、中和小HBsAg,以及羧基端截斷的HBx。在病毒復制及慢性感染維持方面,整合的HBV DNA可能在以下幾個環節參與其中:(1)轉錄表達HBsAg為病毒提供包膜以維持HBV的持續感染;(2)整合來源的HBsAg和羧基端截斷的HBx可能參與cccDNA的轉錄活性調節和病毒復制;(3)在長期的病毒-宿主相互作用下,肝臟中的整合事件通過免疫逃逸和肝細胞克隆性擴增而逐漸增加,而cccDNA池則逐漸丟失或轉錄沉默,最終可能導致整合的HBV DNA成為HBeAg陰性患者血清HBsAg的主要來源。除此之外,整合HBV DNA的持續存在與表達,可能是CHB特別是HBeAg陰性患者難以治愈的主要原因。未來,應進一步加強對HBV整合參與病毒復制及維持慢性感染的作用及相關機制的研究,以進一步提高CHB患者的臨床治愈。
猜你喜歡
中老年保健(2021年3期)2021-08-22 06:50:04
天津醫科大學學報(2021年2期)2021-03-29 05:31:08
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
昆明醫科大學學報(2020年12期)2021-01-26 00:44:04
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
豬業科學(2018年8期)2018-09-28 01:27:38
中成藥(2017年8期)2017-11-22 03:18:47
川北醫學院學報(2015年5期)2015-12-05 08:22:29
