基于UPLC指紋圖譜和多成分定量評價不同產地升麻藥材質量
2022-09-05 05:45:42周湘媛陳萬發馬懿飛曹斯瓊霍文杰李振雨
中草藥 2022年17期
關鍵詞:質量
周湘媛,陳萬發,丁 青,馬懿飛,曹斯瓊,霍文杰,魏 梅,秦 升,李振雨*
基于UPLC指紋圖譜和多成分定量評價不同產地升麻藥材質量
周湘媛1, 2,陳萬發1, 2,丁 青1, 2,馬懿飛1, 2,曹斯瓊1, 2,霍文杰1,魏 梅1, 2,秦 升3,李振雨1, 2*
1. 廣東一方制藥有限公司,廣東 佛山 528244 2. 廣東省中藥配方顆粒企業重點實驗室,廣東 佛山 528244 3. 中國中藥控股有限公司,廣東 佛山 528303
采用超高效液相色譜法(UPLC)建立升麻藥材指紋圖譜,并同時測定3種有效成分的含量,評價不同產地升麻藥材質量的差異。采用UPLC法進行測定,色譜柱為Agilent SB C18(100 mm×2.1 mm,1.8 μm);流動相為乙腈-0.05%磷酸溶液,梯度洗脫,體積流量為0.3 mL/min,波長為320 nm,柱溫為35 ℃,進樣量為1 μL;建立15批升麻藥材指紋圖譜,通過對照品比對并結合質譜分析,對共有峰進行鑒定,并對3種成分的含量進行測定;對15批升麻藥材指紋圖譜進行相似度評價及主成分分析(principal component analysis,PCA),利用正交偏最小二乘法-判別分析(orthogonal partial least squares discriminant analysis,OPLS-DA)尋找不同產地升麻藥材的差異性成分。升麻藥材指紋圖譜有16個共有峰,指認出其中3個峰,分別為咖啡酸、阿魏酸和異阿魏酸;除遼寧產區的3批樣品外,其余12批相似度均大于0.95;PCA將15批升麻藥材劃分為2類,OPLS-DA法確定7個差異標志物,差異顯著性排序,分別為峰7>峰11>峰9>峰8>峰10>咖啡酸>峰4。遼寧產區咖啡酸、阿魏酸和異阿魏酸總含量明顯高于其它產地。該方法能有效地分析不同產地升麻藥材質量的差異性,為不同產地升麻藥材質量的評價提供參考。
升麻藥材;UPLC指紋圖譜;多成分定量;差異標志物;咖啡酸;阿魏酸;異阿魏酸
升麻為毛茛科植物大三葉升麻Kom.、興安升麻(Turcz.) Maxim.或升麻L.的干燥根莖,具有發表透疹、清熱解毒、升舉陽氣之功效,用于治療風熱頭痛、齒痛、口瘡、咽喉腫痛、麻疹不透、陽毒發斑、脫肛、子宮脫垂[1]。已有研究表明,升麻具有抗病毒、抗炎、抗腫瘤、抗骨質疏松、抑制核苷運轉等多種作[2],黃廣欣等[3]研究預測認為,升麻中的酚酸、三萜及其苷類等是升麻藥材中與藥效和傳統藥性相關的主要質量標志物。中藥有效成分的多樣性及復雜性,單一成分定量的化學藥品質量控制模式既缺乏專屬性,又難以反映中藥內在質量屬性,因此,需要創新研究思維和基于功能主治的多元質量控制模式[4],指紋圖譜結合化學計量學對中藥的整體質量控制提供一種的新的思路[5-6],在中藥材質量控制、中藥飲片炮制、中藥復方和中成藥的質量標準研究中屢見不鮮[7-10],但對于采用UPLC指紋圖譜結合化學計量學評價升麻藥材的質量仍鮮有報道。本研究采用UPLC指紋圖譜和主成分分析(principal component analysis,PCA)、正交偏最小二乘法-判別分析(orthogonal partial least squares discriminant analysis,OPLS-DA),并結合質譜指認和多成分定量,從多角度評價不同產地大三葉升麻的質量,為升麻藥材質量標準的提高提供參考。
1 儀器與試藥
1.1 儀器
Waters H-Class型超高效液相色譜儀(美國沃特世公司);Scientific Q-Exactive Obitriap MS型四極桿-靜電軌道阱高分辨質譜儀(美國賽默飛公司);mzVault質譜數據庫(美國賽默飛公司)Agilent SB-C18色譜柱(100 mm×2.1 mm,1.8 μm,美國安捷倫公司),ME204E型萬分之一天平(梅特勒-托利多公司);XP26型百萬分之一天平(梅特勒-托利多公司);JJ600型百分之一天平(常熟市雙杰測試儀器廠);KQ500DE型超聲波清洗器(昆山市超聲儀器有限公司);HWS28型恒溫水浴鍋(上海一恒科技有限公司);超純水系統(Milli-Q級,德國默克股份有限公司)。
1.2 試劑與藥材
除液相用甲醇、乙腈(默克股份有限公司),磷酸、甲酸(天津市科密歐化學試劑有限公司)為色譜級,其余試劑皆為分析純。對照品阿魏酸(批號110773-201614,質量分數以99.0%)、異阿魏酸(批號111698-201103,質量分數以99.2%)、咖啡酸對照品(批號110885-200102,質量分數以100.0%,)均購自中國食品藥品檢定研究院;15批升麻藥材經廣東一方制藥有限公司魏梅主任藥師鑒定均為毛茛科植物大三葉升麻Kom.的干燥根莖,經檢驗,均符合《中國藥典》2020年版升麻藥材項下的規定,藥材來源信息見表1。
表1 15批升麻藥材來源信息
Table 1 Origin information form of 15 batches of Cimicifuga Rhizoma
編號藥材批號產地 S1G1708044河北承德 S2G1708045河北承德 S3G1708046河北承德 S4G1709004遼寧鞍山 S5G1709005遼寧鞍山 S6G1709006遼寧鞍山 S7G1709009吉林通化 S8G1709010吉林通化 S9G1709011吉林通化 S10G1709055內蒙古呼倫貝爾鄂倫 S11G1709056內蒙古呼倫貝爾鄂倫 S12G1709057內蒙古呼倫貝爾鄂倫 S13G1709064黑龍江黑河 S14G1709065黑龍江黑河 S15G1709066黑龍江黑河
2 方法與結果
2.1 升麻藥材UPLC指紋圖譜的建立
2.1.1 色譜條件 色譜柱:Agilent SB C18(100 mm×2.1 mm,1.8 μm)色譜柱;流動相為乙腈(A)-0.05%磷酸溶液(B,質譜分析為0.05%甲酸溶液);梯度洗脫:0~1 min,12% A;1~3 min,12%~18% A;3~6 min,18% A;6~13 min,18%~35% A;13~18 min,35%~90% A;18~19 min,90%~80% A;體積流量為0.30 mL/min;檢測波長為320 nm;柱溫為35 ℃;進樣量為1 μL。
2.1.2 對照品溶液的制備 取阿魏酸、咖啡酸和異阿魏酸對照品適量,加10%乙醇制成含阿魏酸、咖啡酸0.1 mg/mL的混合對照品溶液。
2.1.3 供試品溶液的制備 取升麻藥材粉末(過二號篩)約0.5 g,精密稱定,置具塞錐形瓶中,精密加入稀乙醇25 mL,密塞,稱定質量,超聲處理(功率250 W、頻率40 kHz)30 min,放冷,再稱定質量,用稀乙醇補足減失的質量,搖勻,濾過,取續濾液,即得。
2.1.4 質譜條件 采用電噴霧離子源(HESI),噴霧電壓為3.24 kV,掃描模式為正、負離子模式;掃描范圍為/150~2000;鞘氣體積流量為35 arb;輔助氣體積流量為10 arb;毛細管溫度為350 ℃;輔助器加熱溫度為350 ℃;一級掃描分辨率為70 000 FWHM;二級掃描分辨率為17 500 FWHM;二級碰撞能量為40 eV;S-lens電壓為50 V。
2.1.5 精密度試驗 取升麻藥材(批號G1709055)供試品溶液,按“2.1.1”項下確定的色譜條件下連續進樣6次,檢測指紋圖譜,以異阿魏酸色譜峰為參照峰(S),計算各共有指紋峰與S峰的相對保留時間和相對峰面積RSD值,均小于3.0%。
2.1.6 重復性試驗 取同一批次的升麻藥材粉末(批號G1709055)共計6份,按“2.1.3”項下確定的制備方法制備6份供試品溶液,分別進樣測定,檢測指紋圖譜,以異阿魏酸色譜峰為參照峰(S),計算各共有指紋峰與S峰的相對保留時間和相對峰面積RSD值均小于3.0%。
2.1.7 穩定性試驗 取升麻藥材(批號G1709055)供試品溶液,分別在0、2、4、6、8、12、24 h進樣分析,檢測指紋圖譜,以異阿魏酸色譜峰為參照峰(S),計算各共有指紋峰與S峰的相對保留時間和相對峰面積RSD值均小于3.0%。
2.1.8 指紋圖譜的建立及共有峰標定 取15批升麻藥材樣品,按“2.1.3”項下方法制備15份供試品溶液,分別按“2.1.1”項下確定的色譜條件進行測定,記錄各樣品指紋圖譜的色譜圖,并導出指紋圖譜的.cdf格式,將15批升麻藥材.cdf格式導入“中藥色譜指紋圖譜相似度評價系統(2012版)”軟件中,以編號為S1樣品(批號G1708044)的指紋圖譜為參照圖譜,以6號峰為Mark峰進行保留時間校正,同時進行全峰匹配,15批升麻藥材和升麻對照藥材指紋圖譜疊加圖見圖1,以平均數法生成升麻藥材對照指紋圖譜,見圖2。異阿魏酸為升麻的主要有效成分之一,在所有的已知色譜峰里位置最為居中,且該成分色譜峰峰面積較大,與相鄰的色譜峰分離效果好,另外,該對照品容易獲得,因此選擇異阿魏酸色譜峰為參照峰(S),標定出16個共有指紋峰,計算各指紋峰的相對保留時間和相對峰面積,各指紋峰的相對保留時間RSD值在0.05%~0.33%,相對峰面積RSD值在30.85%~79.35%。
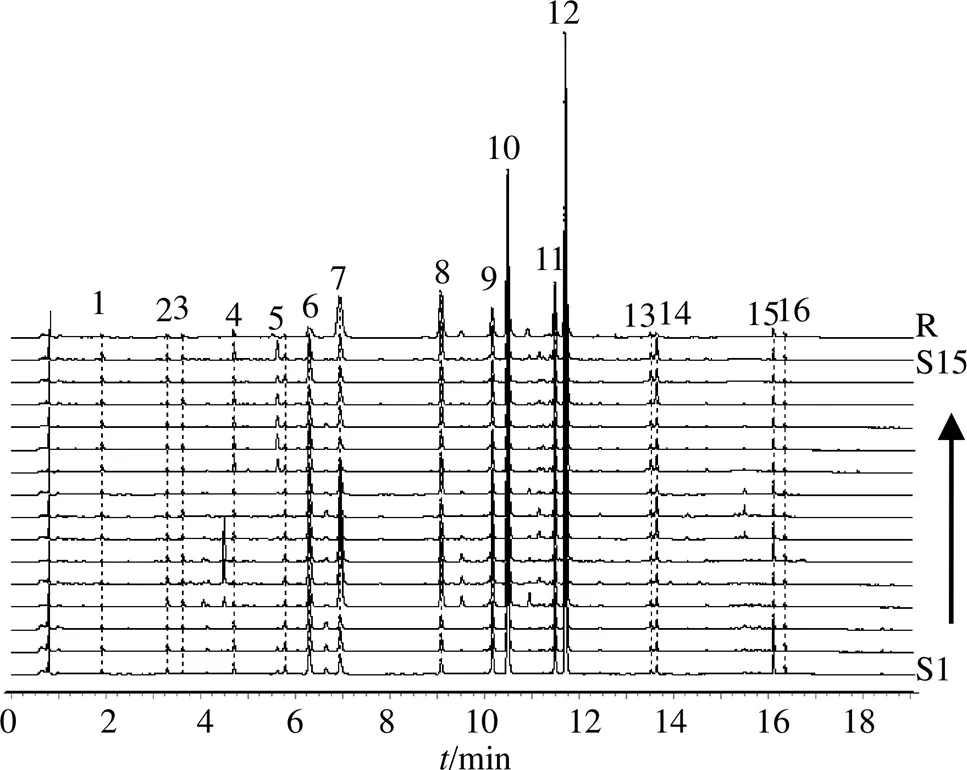
圖1 15批升麻藥材UPLC指紋圖譜
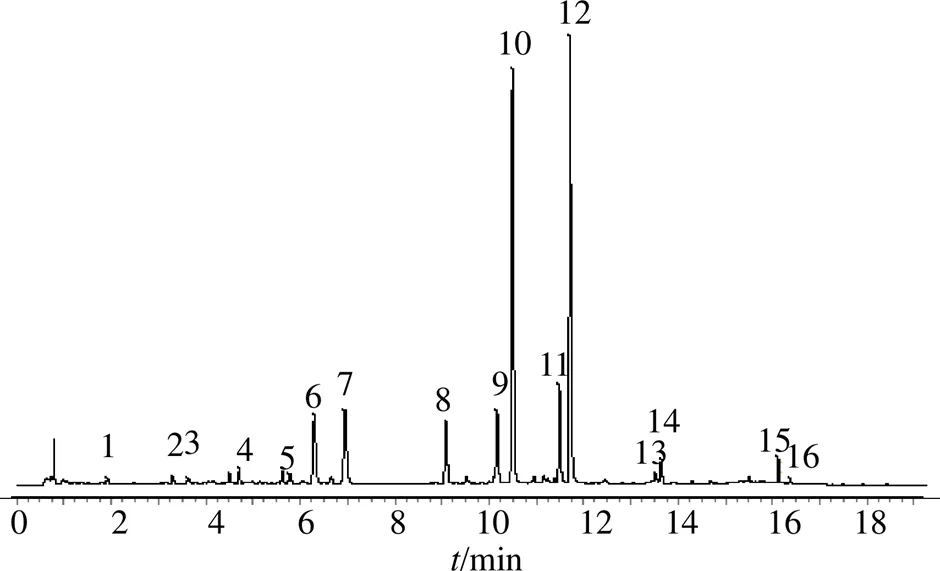
圖2 升麻藥材對照指紋圖譜
2.1.9 相似度評價 采用“中藥色譜指紋圖譜相似度評價系統”計算15批升麻藥材指紋圖譜的相似度(表2),結果顯示,以省劃分產區,不同產區的升麻藥材除編號為S4~S6的藥材外,其余12批升麻藥材指紋圖譜與對照指紋圖譜的相似度均大于0.95,說明河北、吉林、內蒙古和黑龍江4個產區的升麻藥材質量比較一致,遼寧的3批與其他產地相比具有一定的差異。
表2 15批升麻藥材相似度評價
Table 2 Similarity analysis forms of 15 batches of Cimicifuga Rhizoma
相似度S1S2S3S4S5S6S7S8S9S10S11S12S13S14S15對照指紋圖譜 S11.000 1.000 0.994 0.977 0.879 0.954 0.991 0.994 0.971 0.936 0.947 0.929 0.938 0.971 0.940 0.989 S21.000 1.000 0.994 0.978 0.879 0.955 0.990 0.994 0.970 0.935 0.946 0.928 0.938 0.970 0.939 0.989 S30.994 0.994 1.000 0.966 0.861 0.936 0.993 0.998 0.971 0.947 0.956 0.934 0.941 0.983 0.939 0.986 S40.977 0.978 0.966 1.000 0.926 0.981 0.968 0.969 0.969 0.905 0.923 0.913 0.923 0.948 0.927 0.935 S50.879 0.879 0.861 0.926 1.000 0.971 0.840 0.854 0.855 0.738 0.753 0.740 0.732 0.819 0.764 0.875 S60.954 0.955 0.936 0.981 0.971 1.000 0.923 0.938 0.916 0.827 0.847 0.829 0.842 0.892 0.848 0.944 S70.991 0.990 0.993 0.968 0.840 0.923 1.000 0.993 0.986 0.969 0.977 0.964 0.968 0.990 0.971 0.995 S80.994 0.994 0.998 0.969 0.854 0.938 0.993 1.000 0.968 0.944 0.955 0.933 0.947 0.978 0.939 0.986 S90.971 0.970 0.971 0.969 0.855 0.916 0.986 0.968 1.000 0.971 0.978 0.976 0.964 0.989 0.984 0.993 S100.936 0.935 0.947 0.905 0.738 0.827 0.969 0.944 0.971 1.000 0.998 0.994 0.986 0.986 0.982 0.963 S110.947 0.946 0.956 0.923 0.753 0.847 0.977 0.955 0.978 0.998 1.000 0.995 0.992 0.989 0.985 0.973 S120.929 0.928 0.934 0.913 0.740 0.829 0.964 0.933 0.976 0.994 0.995 1.000 0.990 0.977 0.992 0.964 S130.938 0.938 0.941 0.923 0.732 0.842 0.968 0.947 0.964 0.986 0.992 0.990 1.000 0.971 0.979 0.966 S140.971 0.970 0.983 0.948 0.819 0.892 0.990 0.978 0.989 0.986 0.989 0.977 0.971 1.000 0.975 0.987 S150.940 0.939 0.939 0.927 0.764 0.848 0.971 0.939 0.984 0.982 0.985 0.992 0.979 0.975 1.000 0.971 對照指紋圖譜0.989 0.989 0.986 0.9350.875 0.944 0.995 0.986 0.993 0.963 0.973 0.964 0.966 0.987 0.971 1.000
2.1.10 共有峰的指認 采用與對照品的保留時間并結合紫外吸收光譜圖,初步確定峰2為咖啡酸,峰5為阿魏酸,峰6為異阿魏酸,結果見圖3。取升麻藥材(批號G1709055)供試品溶液,按“2.1.1”項下色譜條件和“2.1.4”項下質譜條件進樣分析,根據高分辨質譜提供的準確分子量與升麻已知組分的分子量進行對比,對于質量偏差小于2×10?9的組份予以確認,且根據目標色譜峰的二級質譜信息,并與mzVault標準數據庫進行匹配進行進一步確認,結果見表3。
2.2 化學模式識別
化學計量學中的模式識別技術已廣泛應用于中藥材產地、基原、炮制、真偽鑒別等方面[11],分為有、無監督2種統計方法,一般先進行無監督的HCA、PCA,觀察樣本之間是否有分類趨勢,再采用PLS-DA或者OPLS-DA進行有監督的模式識別,顯示樣本間差異主要由哪些變量引起,尋找差異性標志物[12],

圖3 對照品咖啡酸(a)、阿魏酸(b)、異阿魏酸(c) 及升麻藥材樣品色譜圖(d)
表3 升麻藥材共有峰的質譜信息
Table 3 Mass spectrum information of common peak of Cimicifuga Rhizoma
峰號tR/min分子式分子離子峰實驗值(m/z)理論值(m/z)誤差(×10?6)化合物 23.39C9H8O4[M?H]?179.034180.1570.641咖啡酸 56.01C10H10O4 [M+H]+195.066194.0581.203阿魏酸 66.52C10H10O4 [M+H]+195.065194.0580.947 異阿魏酸
2.2.1 PCA 采用SIMCA14.1軟件,以15批升麻藥材的16個共有指紋峰的峰面積為變量進行PCA分析,共生成9個主成分,累計貢獻率達到99.2%,2為0.56,證明模型有效。以前2個主成分的得分值和16個共有指紋峰的峰面積為變量得到主成分得分圖(Scores圖,圖4)和變量載荷圖(Loading圖,圖5),結果顯示,15批升麻藥材可分為2類,河北、吉林、內蒙古和黑龍江4個產區的升麻藥材聚為一類,遼寧產區升麻聚為第2類,分類結果與藥材產地相關性較大。根據變量離原點的距離判斷變量對主成分的影響權重,距離原點越遠,變量對主成分的影響權重越大,分析變量對主成分1的影響權重,大小依次為峰9、峰7、咖啡酸,變量對主成分2的影響權重,大小依次為峰8、阿魏酸、峰11。
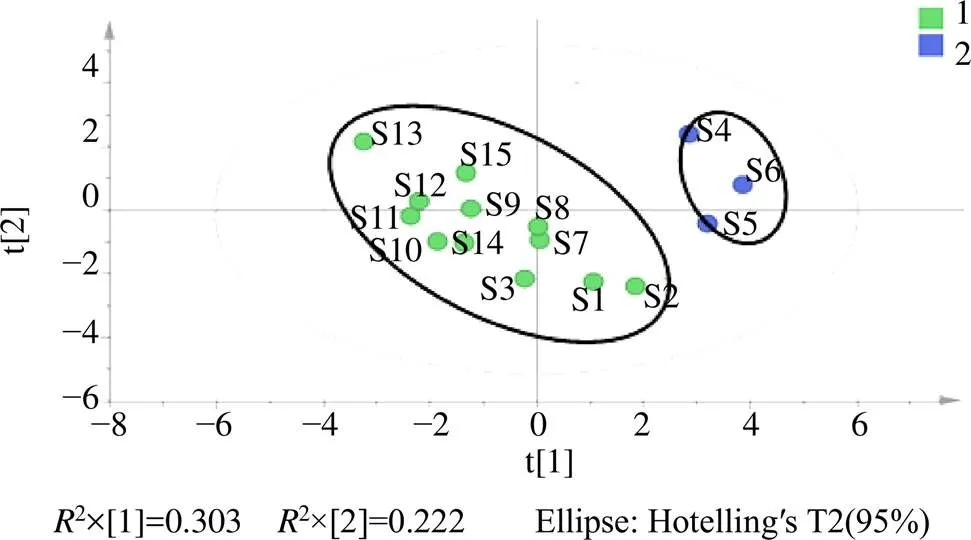
圖4 15批升麻藥材Scores圖
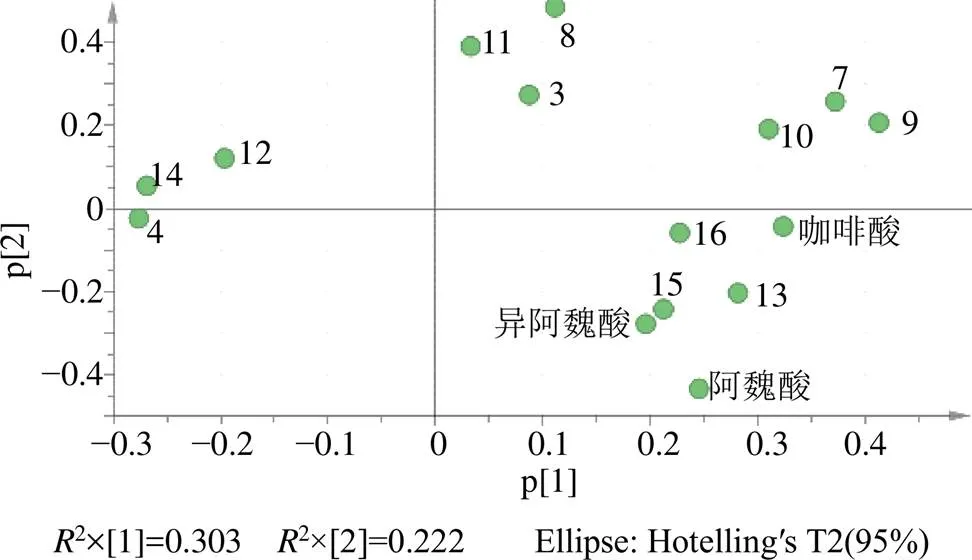
圖5 升麻藥材色譜峰變量Loading圖
2.2.2 OPLS-DA 根據PCA分析結果,以16個共有指紋峰峰面積為變量進行OPLS-DA,所建立的模型2=0.916,2=0.967,2=0.776,表明該模型可以用于不同產地升麻的模式識別。通過OPLS-DA的Scores圖(圖6)可知,不同產地升麻藥材也明顯分為3類,且通過16個變量的VIP值圖(圖7)可知,以VIP>1.0為顯著影響,共找到7個差異標志物,對其影響顯著性排序,分別為峰7>峰11>峰9>峰8>峰10>咖啡酸>峰4,提示這幾個化學成分對于區分不同產地升麻的貢獻較大,且不同產地的咖啡酸含量具有明顯區別。
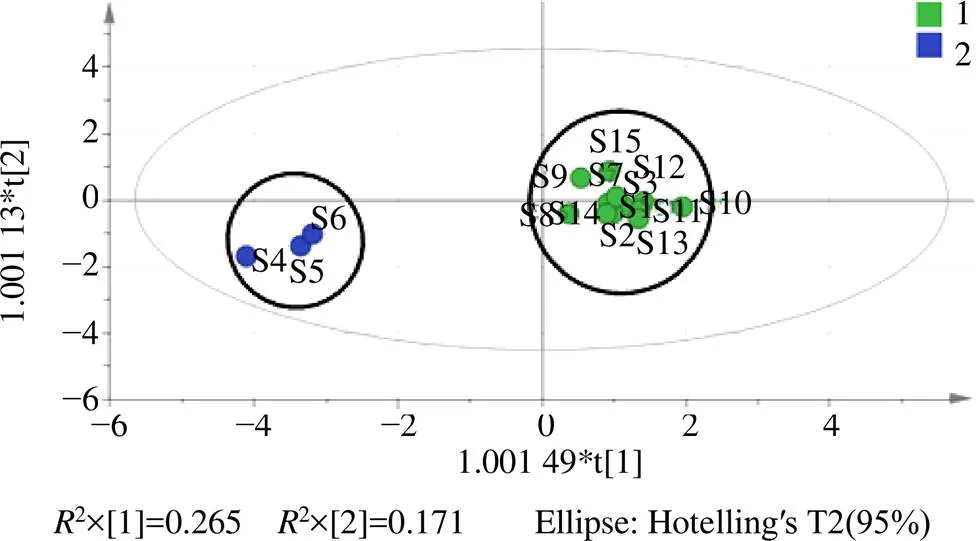
圖6 升麻藥材的OPLS-DA分析Scores圖
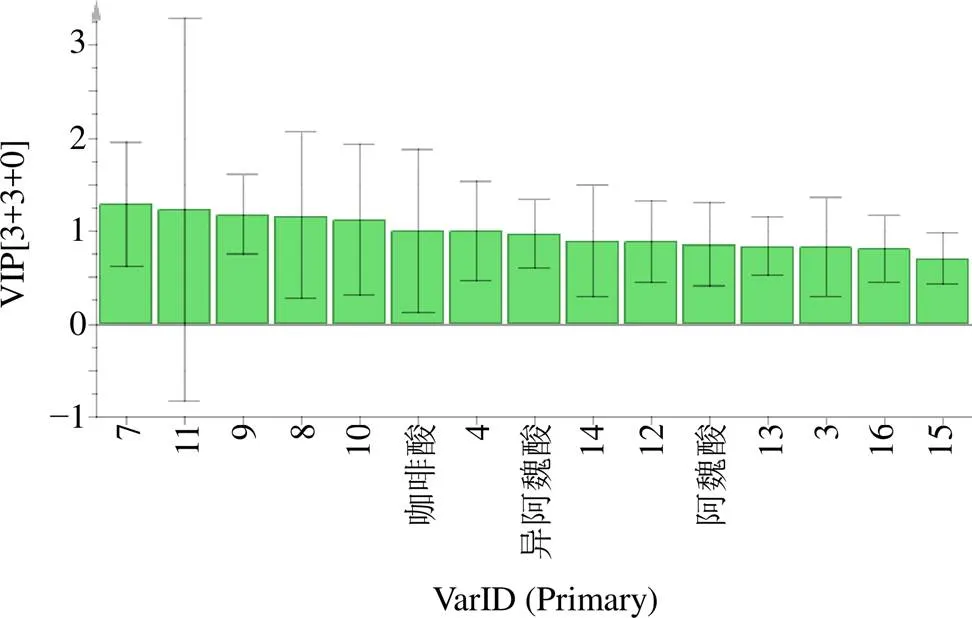
圖7 升麻藥材VIP圖
2.3 3種指標成分的含量測定
2.3.1 供試品溶液制備 取升麻藥材粉末(過二號篩)約0.5 g,精密稱定,置具塞錐形瓶中,精密加入10%乙醇25 mL,密塞,稱定質量,加熱回流2.5 h,放冷,再稱定質量,用10%乙醇補足減失的質量,搖勻,濾過,取續濾液,即得。
2.3.2 線性關系考察 取咖啡酸、阿魏酸及異阿魏酸對照品適量,精密稱定,加10%乙醇制成含咖啡酸30.08 μg/mL,阿魏酸59.76 μg/mL,異阿魏酸566.43 μg/mL的混合對照品儲備液。精密移取上述混合對照品儲備液0.1、0.2、0.5、1.0、2.0、5.0 mL,分別置10 mL量瓶中,加10%乙醇至刻度,搖勻,得到分別含咖啡酸0.30、0.60、1.50、3.01、6.02、15.04 μg/mL,阿魏酸0.60、1.20、2.99、5.98、11.95、29.88 μg/mL,異阿魏酸5.66、11.33、28.32、56.64、113.29、283.22 μg/mL的系列對照品溶液,分別精密吸取上述6個不同質量濃度的對照品溶液各1 μL,按“2.1.1”項下色譜條件進樣測定,以色譜峰峰面積為縱坐標(),對照品濃度為橫坐標()進行線性回歸,繪制標準曲線,3種成分的線性回歸方程和線性范圍見表4。
表4 3種成分的線性回歸方程及線性范圍
Table 4 Regression equations coefficient correlation of references and linear ranges
對照品線性范圍/(μg·mL?1)標準曲線R2 咖啡酸0.30~30.08Y=16.40 X+0.640.999 9 阿魏酸0.60~59.76Y=16.41 X+4.150.999 5 異阿魏酸 5.66~566.43Y=16.13 X+6.940.999 9
2.3.3 精密度試驗 取“2.1.2”項下對照品溶液,按“2.1.1”項下色譜方法連續進樣6次,記錄咖啡酸、阿魏酸和異阿魏酸的峰面積,并計算咖啡酸、阿魏酸和異阿魏酸峰面積RSD值分別為0.68%、1.05%、0.89%。
2.3.4 穩定性試驗 取升麻藥材粉末(批號G1709055),按“2.3.1”項下確定的供試品溶液制備方法制備供試品溶液,按“2.1.1”項下色譜條件分別在0、2、4、6、8、10、12、24 h進樣分析,記錄咖啡酸、阿魏酸和異阿魏酸的峰面積,并計算不同時間點咖啡酸、阿魏酸和異阿魏酸峰面積RSD值,分別為0.64%、1.15%、1.10%。
2.3.5 重復性試驗 取升麻藥材粉末(批號G1709055),按“2.3.1”項下確定的供試品溶液制備方法制備6份供試品溶液,并按“2.1.1”項下色譜條件進樣測定,計算咖啡酸、阿魏酸和異阿魏酸的質量分數分別為0.14、0.29、2.83 mg/g,RSD值分別為1.71%、1.27%、0.37%。
2.3.6 加樣回收率測定 取升麻藥材粉末(批號G1709055)約0.25 g,精密稱定,平行3組,每組3份,按1∶0.5,1∶1及1∶1.5的比例加入對照品,按“2.3.1”項下方法制備9份供試品溶液,按“2.1.1”項下色譜條件進樣測定,計算3種成分的回收率范圍及RSD值,計算咖啡酸、阿魏酸和異阿魏酸的加樣回收率范圍分別為92.21%~100.37%、96.82%~101.06%、96.76%~101.63%,平均加樣回收率分別為96.89%、99.14%、99.40%,RSD值分別為2.51%、1.65%、1.70%,均小于3.0%。
2.3.7 中間精密度試驗 由其他分析人員在不同日期和不同色譜儀下操作,取同一批升麻藥材粉末(批號G1709055)約0.5 g,精密稱定,平行6份,按“2.3.1”項下方法制備6份供試品溶液,按“2.1.1”項下色譜條件進樣測定,計算咖啡酸、阿魏酸和異阿魏酸的質量分數分別為0.14、0.30、2.87 mg/g,與6份重復性試驗樣品的RSD值分別為1.77%、2.20%和0.65%。
2.3.8 樣品測定 取15批升麻藥材粉末,按“2.3.1”項下方法制備供試品溶液,按“2.1.1”項下色譜條件進樣測定,記錄色譜圖,按外標法計算各批次樣品咖啡酸、阿魏酸和異阿魏酸的含量,結果見表5,15批升麻藥材咖啡酸質量分數在0.14~0.92 mg/g,阿魏酸質量分數在0.29~0.67 mg/g,異阿魏酸質量分數在2.36~4.03 mg/g,《中國藥典》2020年版升麻藥材項下規定異阿魏酸含量不得少于0.1%,15批樣品均藥典符合規定。遼寧產區的3種成分的整體含量高于其他產地,其余產地3種有效成分的含量無明顯差異,見圖8。
表5 樣品測定結果(n=15)
Table 5 Determination results of samples (n=15)
批號咖啡酸/(mg·g?1)阿魏酸/(mg·g?1)異阿魏酸/(mg·g?1)總量/(mg·g?1) S10.250.442.513.20 S20.210.332.362.90 S30.160.402.923.48 S40.920.564.035.51 S50.870.523.935.32 S60.350.673.714.73 S70.160.472.923.55 S80.170.373.524.06 S90.170.363.453.98 S100.140.292.833.26 S110.150.302.993.44 S120.150.302.943.39 S130.210.432.873.51 S140.210.432.873.51 S150.330.422.563.31
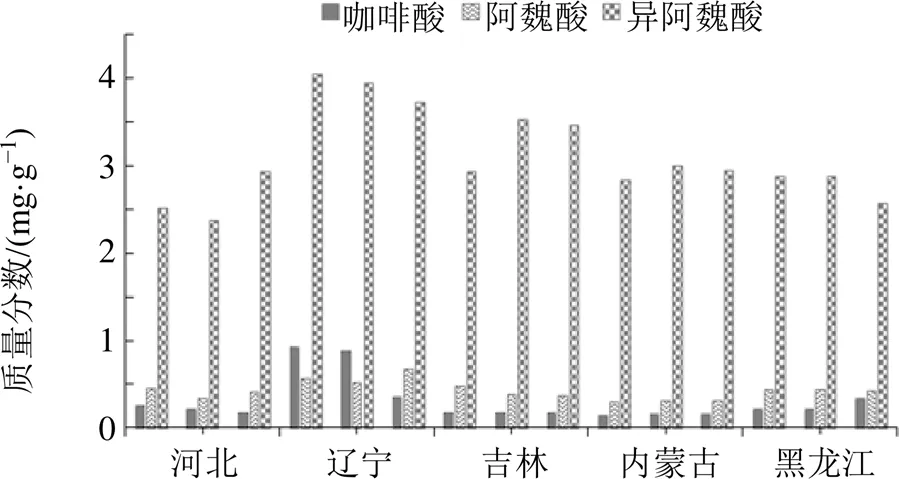
圖8 不同產地間升麻藥材各指標含量均值圖
3 討論
3.1 色譜條件的選擇
本實驗采用UPLC法建立升麻藥材指紋圖譜與有效成分含量同時測定方法,在20 min內即完成主要色譜峰的良好分離,指紋圖譜研究發現,320 nm處色譜峰數目較多,響應值較大,基線平穩,其中咖啡酸、阿魏酸和異阿魏酸3種有效成分的分離度和峰純度符合定量要求,因此選擇320 nm作為檢測波長。流動相的篩選考察了甲醇-0.1%磷酸溶液,乙腈-0.1%磷酸溶液和甲醇-水3種溶劑系統的洗脫和分離效果,結果顯示,以乙腈-0.1%磷酸溶液為流動相,各色譜峰出峰時間短,分離度較好。色譜柱篩選考察了Agilent SBC18(100 mm×2.1 mm,1.8 μm)色譜柱、Waters HSS T3 C18(100 mm×2.1 mm,1.8 μm)色譜柱、Waters Acquity BEH C18(100 mm×2.1 mm,1.7 μm)色譜柱這3種不同品牌和柱填料的色譜柱對各色譜峰的分離效果,結果采用Agilent SB-C18(100 mm×2.1 mm,1.8 μm)色譜柱,各色譜峰分離效果最佳。
3.2 指紋圖譜的構建及分析評價
本研究15批升麻藥材指紋圖譜共標識出16個共有指紋峰,通過對照品指認出3個有效成分,分別為咖啡酸、阿魏酸和異阿魏酸。相似度評價結果顯示,除3批吉林產區的樣品外,其余產地升麻指紋圖譜相似度較高。通過PCA將15批升麻藥材分為2類,其中遼寧產區分為一類,其余產區為一類。通過OPLS-DA分析找到7個差異標志物,說明不同產區升麻藥材的化學成分含量存在一定差異,這種差異可能與藥材的生長環境、生長年限、采收加工方式等因素密切相關。
3.3 有效成分的測定與分析
《中國藥典》2020年版升麻藥材項下以異阿魏酸為含測指標,但僅用單一成分測定尚不足以全面評價升麻藥材的質量,故建立多成分同時測定是實現中藥材有效質控的重要方法。本研究在指紋圖譜的基礎上,增加對有效成分咖啡酸、阿魏酸和異阿魏酸的同時測定,從多角度研究不同產地升麻藥材質量的差異性,結果表明,除遼寧產區外,其余4個產地的咖啡酸、阿魏酸和異阿魏酸含量差異不大,最小值和最大值不超過3倍,其中以遼寧的升麻整體質量較優。
本研究建立升麻指紋圖譜方法及多成分定量,該方法簡便,專屬性強,重復性良好,分析時間短,提高了分析效率,能有效地分析不同產地升麻藥材質量的差異性,為不同產地升麻藥材的質量評價提供參考。
利益沖突 所有作者均聲明不存在利益沖突
[1] 中國藥典 [S]. 一部. 2020: 75.
[2] 孫啟泉, 左愛俠, 張婷婷. 升麻屬植物化學成分、生物活性及臨床應用研究進展 [J]. 中草藥, 2017, 48(14): 3005-3016.
[3] 黃廣欣, 龔蘇曉, 許浚, 等. 升麻研究進展及其質量標志物的預測分析 [J]. 中草藥, 2020, 51(10): 2651-2660.
[4] 鄢海燕, 鄒純才. 基于功能主治及其物質基礎的中藥多元化質量控制模式探討 [J]. 國際藥學研究雜志, 2019, 46(4): 266-269.
[5] 劉東方, 趙麗娜, 李銀峰, 等. 中藥指紋圖譜技術的研究進展及應用 [J]. 中草藥, 2016, 47(22): 4085-4094.
[6] 于洋, 李軍, 李寶國. 化學計量學在中藥質量控制研究中的應用 [J]. 中成藥, 2018, 40(5): 1139-1142.
[7] 陶曉賽, 龔海燕, 謝彩俠, 等. 基于UPLC指紋圖譜結合化學計量學評價不同產地盾葉薯蕷藥材質量 [J]. 中草藥, 2021, 52(1): 227-233.
[8] 張曉男, 魏惠珍, 張丹, 等. 基于化學計量學分析的桃仁炮制前后指紋圖譜研究 [J]. 中國現代應用藥學, 2020, 37(8): 971-976.
[9] 周麗, 時海燕, 時銀萍, 等. 指紋圖譜結合化學計量學優選經典名方溫膽湯的提取工藝 [J]. 中國醫院藥學雜志, 2020, 40(21): 2214-2219.
[10] 高森, 王蘋, 唐鋮, 等. 基于HPLC指紋圖譜、多指標成分含量測定及化學計量學的濕熱痹片質量評價 [J]. 中草藥, 2020, 51(21): 5454-5461.
[11] 孫立麗, 王萌, 任曉亮. 化學模式識別方法在中藥質量控制研究中的應用進展 [J]. 中草藥, 2017, 48(20): 4339-4345.
[12] 于洋, 李軍, 李寶國. 化學計量學在中藥質量控制研究中的應用 [J]. 中成藥, 2018, 40(5): 1139-1142.
Quality evaluation offrom different producing areas based on fingerprint and multi-components determination
ZHOU Xiang-yuan1, 2, CHEN Wan-fa1, 2, DING Qing1, 2, MA Yi-fei1, 2, CAO Si-qiong1, 2, HUO Wen-jie1, WEI Mei1, 2, QIN Sheng3, LI Zhen-yu1, 2
1. Guangdong E-fong Pharmaceutical Co. Ltd, Foshan 528244, China 2. Guangdong Provincial Key Laboratory of Traditional Chinese Medicine Formula, Foshan 528244, China 3. China Traditional Chinese Medicine Holdings Co. Ltd., Foshan 528303, China
To evaluate the difference offrom different producing areas by establishing UPLC fingerprint and determining contents of three effective components ofUPLC Method was adopted. The determination was performed on a column of Agilent SB C18(100 mm × 2.1 mm,1.9 μm) with acetonitrile-0.05% phosphoric acid solution as the mobile phase by gradient elution at a flow rate of 0.3 mL/min. The detective wavelength was 320 nm, the column temperature was 35 ℃, the injection volume was 1 μL. The UPLC fingerprints of 15 batches ofwere established and the common peaks were identificated by reference substances and mass spectrometry. The contents of three components were determined. Similarity evaluation and principal component analysis (PCA) were carried out on the fingerprints and orthogonal partial least squares discriminant analysis (OPLS-DA) was used to find the different components offrom different producing areas.There were 16 common peaks in the fingerprints ofby comparing with the reference substances, three common peaks were identified, namely, caffeic acid, ferulic acid and isoferulic acid. Except for three batches of samples from Liaoning Province, the similarity of the other 12 batch was greater than 0.95. According to the analysis of PCA, 15 batches ofwere divided into two categories; Seven kinds of biomarkers were determined by OPLS-DA method. The order of significance was peak 7 > peak 11 > peak 9 > peak 8 > peak 10 > caffeic acid > peak 4, respectively. The total contents of caffeic acid, ferulic acid and isoferulic acid.in Liaoning were obviously higher than those in other areas.This method can effectively analyze the quality differences offrom different producing areas and provide reference for the quality evaluation.
; UPLC fingerprints; multi-component determination; difference markers; caffeic acid; ferulic acid; isoferulic acid
R286.2
A
0253 - 2670(2022)17 - 5497 - 07
10.7501/j.issn.0253-2670.2022.17.027
2022-01-20
廣東省省級科技計劃項目(2018B030323004);廣東特支計劃科技創業領軍人才項目(2017TY04R197)
周湘媛(1996—),女,助理研究員,從事中藥飲片及中藥配方顆粒研究。
李振雨(1989—),男,碩士,主管中藥師,研究員,從事中藥飲片及中藥配方顆粒研究。Tel: (0757)85128602 E-mail: 1083656123@qq.com
[責任編輯 時圣明]
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
