HPLC雙標多測法測定銀黃口服液中7個成分的含量
2022-08-08 03:21:02欒永福周廣濤許麗麗馬雙成林永強郭東曉
食品與藥品 2022年4期
欒永福,周廣濤,許麗麗, ,馬雙成,孫 磊,林永強*,郭東曉*
(1. 山東省食品藥品檢驗研究院 國家藥監局膠類產品質量評價重點實驗室 山東省中藥標準創新與質量評價工程實驗室,山東 濟南 250101;2. 山東中醫藥大學,山東 濟南 250355;3. 中國食品藥品檢定研究院,北京 100050)
定性和定量是藥物質量評價和控制的主要技術手段,而標準物質則是成分定性和定量研究的關鍵因素。標準物質存在提純困難、價格高昂、不穩定或毒性大等問題,尤其隨著行業的快速發展,標準物質需求也越來越大,這些都對標準物質的供應和使用提出了更高的要求。中藥成分復雜,采用多指標分析測定技術才能全面地進行藥物質量評價,標準物質的短缺等因素限制了該技術的應用。為解決這一問題,目前研究使用最多的是一測多評法[1-6],但該方法的色譜柱耐用性較差[7],制約了該方法的應用和推廣。雙標多測法[8-10]是近年提出的一種新的替代對照品法,采用雙標線性校正法進行定性,其原理是以兩個標準物質為參照預測其他成分的保留時間,然后以雙標化合物中一個標準物質為參照物,采用最大吸收波長相對校正因子法進行定量。與一測多評法相比,雙標多測法在儀器、色譜柱的耐用性和定性定量準確度方面均有一定的優勢,目前已經在藥物分析檢測領域有了較廣泛、深入的研究和應用[11-14]。此外,目前已經建立了科學、系統、完善的軟件分析工具──數字標準物質平臺(DRS Origin),保證雙標線性校正法的科學計算與應用。
銀黃口服液由金銀花提取物和黃芩提取物組方而成,具有清熱疏風、利咽解毒之功效[15],方中主要成分為6種咖啡酰奎寧酸和黃芩苷等。《中華人民共和國藥典》2020年版一部項下,只對綠原酸和黃芩苷進行含量控制,指標單一,難以對質量進行有效控制。由于除綠原酸和黃芩苷之外的其他成分的標準物質價格較昂貴,因此本實驗以DRS Origin軟件為平臺,采用雙標多測法測定銀黃口服液中6種咖啡酰奎寧酸和黃芩苷的含量,驗證該方法在銀黃口服液質量評價和控制中的可行性。
1 儀器與材料
1.1 儀器
安捷倫1200高效液相色譜儀(配紫外檢測器,美國安捷倫);Sartorius CP225D電子天平(德國Sartorius公司);Millipore Q-POD?超純水處理儀(美國Millipore公司);26根色譜柱信息見表1。

表1 色譜柱信息表
1.2 材料
綠原酸對照品(批號:110753-201415,含量:96.2 %),3, 5-O-二咖啡酰奎寧酸對照品(批號:111782-201807,含量:94.3 %),4, 5-O-二咖啡酰奎寧酸對照品(批號:111894-201102,含量:94.1 %),黃芩苷對照品(批號:110715-201318,含量:93.3 %,中國食品藥品檢定研究院);新綠原酸對照品(批號:MUST-19031001,含量:99.67 %),隱綠原酸(批號:MUST-19032403,含量:99.07 %),3, 4-O-二咖啡酰奎寧酸(批號:MUST-19031602,含量:99.05 %,成都曼斯特生物科技有限公司)。乙腈,磷酸(色譜純,Merk公司),其余試劑為分析純。20批銀黃口服液樣品來源于不同生產企業,詳細信息見表2。

表2 樣品信息
1.3 軟件
數字化標準物質大數據平臺(DRS Origin)V1.0(中國食品藥品檢定研究院)。
2 方法與結果
2.1 色譜條件[15]
色譜柱:以十八烷基硅烷鍵合硅膠為填充劑;流動相:乙腈(A)-0.4 %磷酸溶液(B),梯度洗脫(0~15 min,5 %A→20 %A;15~30 min,20 %A→30 %A;30~40 min,30 %A);檢測波長:327 nm;流速:1.0 ml/min,柱溫:35 ℃;進樣量:10 μl。
2.2 對照品溶液的制備
精密稱取新綠原酸對照品11.38 mg、綠原酸對照品11.38 mg、隱綠原酸對照品18.40 mg、3, 4-O-二咖啡酰奎寧酸對照品11.02 mg、3, 5-O-二咖啡酰奎寧酸對照品10.74 mg、4, 5-O-二咖啡酰奎寧酸對照品10.67 mg、黃芩苷對照品23.24 mg,分別置25,25,50,25,25,25,50 ml棕色量瓶中,加50 %甲醇使溶解并稀釋至刻度,搖勻,作為對照品儲備液;分別精密量取上述對照品儲備液各5 ml,置同一100 ml棕色量瓶中,加50 %甲醇至刻度,搖勻,即得每1 ml各含22.68,21.90,18.23,21.83,20.26,20.08,22.17 μg的混合對照品溶液,即得。
2.3 供試品溶液的制備
精密量取本品1 ml,置50 ml棕色量瓶中,加50 %甲醇稀釋至刻度,搖勻,濾過,取續濾液,即得。
2.4 定性研究
2.4.1 色譜柱的篩選 在色譜柱1~25上采集對照品溶液和供試品溶液的色譜圖,其中在色譜柱1、色譜柱21、色譜柱23上,綠原酸和隱綠原酸峰均未達到完全分離,故以其余22根色譜柱上7個成分的保留時間進行統計分析。
2.4.2 雙標化合物的選擇 將對照品溶液在上述22根色譜柱上的色譜圖導入DRS Origin軟件,以7個成分中任意2種成分在上述22根色譜柱上得到的保留時間平均值[標準保留時間(standard retention time,SRT)]為橫坐標,以其在每根色譜柱上的實際保留時間為縱坐標,得到7個成分在每根色譜柱上的線性擬合方程,將其他5種成分的SRT代入方程,求得7個成分在每根色譜柱上的預測保留時間。根據DRS Origin軟件中各色譜峰保留時間回歸偏差、預測正確率和色譜柱符合率情況,按照雙標選擇盡量分布在保留時間區域的兩端的原則,同時兼顧對照品易得程度,選擇峰2(綠原酸)和峰7(黃芩苷)作為雙標化合物。
2.4.3 雙標線性校正法的建立 在色譜柱26上采集雙標化合物(綠原酸和黃芩苷)色譜峰實際保留時間作為縱坐標,以二者的SRT為橫坐標,得到雙標化合物在色譜柱26上的線性擬合方程為y=0.9631x-0.0865,即建立雙標線性校正法。
2.4.4 保留時間的預測與結果驗證 將其他5種成分的SRT代入此方程,求得新綠原酸、隱綠原酸、3,4-O-二咖啡酰奎寧酸、3, 5-O-二咖啡酰奎寧酸、4,5-O-二咖啡酰奎寧酸在色譜柱26上的預測保留時間。經實物對照品驗證,5種成分的預測保留時間與實測保留時間的誤差不超過0.5 min,結果準確,見表3。可見使用DRS Origin軟件建立雙標線性校正法,方法簡便科學。

表3 色譜柱26上7個成分的實測保留時間和預測保留時間
2.5 定量研究
2.5.1 典型色譜圖 在色譜柱26上,進行上述7個成分的定量研究,典型色譜圖見圖1。
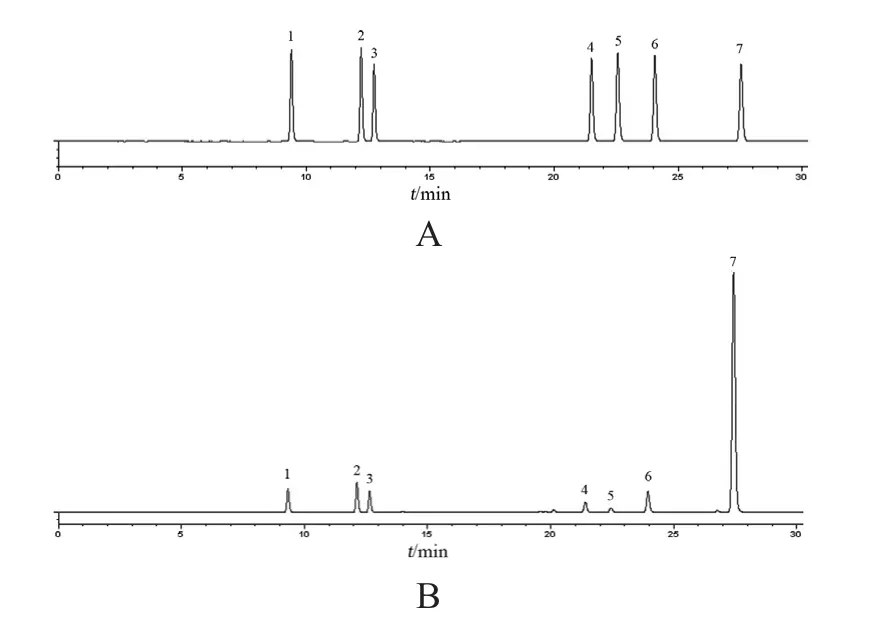
圖1 混合對照品和供試品的HPLC圖譜
2.5.2 檢測波長的確定 新綠原酸、綠原酸、隱綠原酸、3, 4-O-二咖啡酰奎寧酸、3, 5-O-二咖啡酰奎寧酸、4, 5-O-二咖啡酰奎寧酸均在327±2 nm處有最大吸收,而黃芩苷在278±2 nm處有最大吸收,故選擇以327 nm作為新綠原酸、綠原酸、隱綠原酸、3,4-O-二咖啡酰奎寧酸、3, 5-O-二咖啡酰奎寧酸、4,5-O-二咖啡酰奎寧酸的檢測波長,以278 nm作為黃芩苷的檢測波長。
2.5.3 相對校正因子的計算 將2.2項下的各對照品儲備液分別稀釋250,100,50,25,20,10倍,制成混合對照品系列稀釋液,精密吸取10 μl,注入液相色譜儀,依前述色譜條件進行分析,以進樣量對峰面積進行回歸,分別得到其余6種成分的相對校正因子(f)。
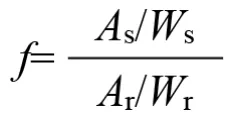
式中,As、Ar分別為待測組分和參照物的峰面積;Ws、Wr分別為待測組分和參照物的進樣量。根據公式,求得新綠原酸、隱綠原酸、3, 4-O-二咖啡酰奎寧酸、3, 5-O-二咖啡酰奎寧酸、4, 5-O-二咖啡酰奎寧酸和黃芩苷對綠原酸的f值分別為0.9606,09612,1.0243,1.2603,1.2394,1.0872。
2.5.4 方法學考察
2.5.4.1 精密度試驗 精密吸取2.2項下的混合對照品溶液10 μl,注入液相色譜儀,連續進樣6次,對7個對照品色譜峰的保留時間和峰面積進行統計,結果表明各對照品的保留時間和峰面積積分值的RSD均小于2.0 %,符合分析要求。
2.5.4.2 線性關系考察 分別精密吸取2.5.3項下的混合對照品系列稀釋液和2.2項下黃芩苷對照品儲備液各10 μl,注入液相色譜儀,測定峰面積積分值。以各對照品的進樣量為橫坐標,峰面積積分值為縱坐標,進行線性回歸,得回歸方程。結果表明,新綠原酸、綠原酸、隱綠原酸、3, 4-O-二咖啡酰奎寧酸、3, 5-O-二咖啡酰奎寧酸、4, 5-O-二咖啡酰奎寧酸和黃芩苷進樣量分別在18.15~453.70 ng、17.52~437.90 ng、16.50~412.53 ng、17.46~436.61 ng、15.19~379.84 ng、15.45~386.19 ng、17.73~4433.62 ng范圍內和峰面積積分值呈良好的線性關系。
2.5.4.3 重復性試驗 取樣品1,按2.3項下方法制備供試品溶液,平行制備6份。分別精密吸取2.2項下混合對照品溶液及上述供試品溶液各10 μl,注入液相色譜儀,測定,對7個成分色譜峰的保留時間和峰面積積分值進行統計。結果表明各對照品的保留時間和峰面積積分值的RSD均小于2.0 %,符合分析要求。
2.5.4.4 穩定性試驗 取樣品1,按2.3項下方法制成供試品溶液,每隔4 h進樣一次,每次10 μl,測定7個成分的峰面積積分值,共考察24 h,以觀察供試品溶液在檢測過程中待測成分的穩定性。結果表明各對照品的保留時間和峰面積積分值的RSD均小于2.0 %,符合分析要求。
2.5.4.5 加樣回收試驗 取已測知含量的樣品1,精密量取0.5 ml,置50 ml棕色量瓶中,分別精密加入新綠原酸、綠原酸、隱綠原酸、3, 4-O-二咖啡酰奎寧酸、3, 5-O-二咖啡酰奎寧酸,4, 5-O-二咖啡酰奎寧酸和黃芩苷對照品儲備液0.75,1,1,0.5,0.2,1,20 ml,再加50 %甲醇稀釋至刻度,搖勻,濾過,取續濾液,即得。平行制備6份。分別精密吸取2.2項下對照品溶液及上述供試品溶液各10 μl,注入液相色譜儀,測定,計算含量。新綠原酸,綠原酸,隱綠原酸,3, 4-O-二咖啡酰奎寧酸,3, 5-O-二咖啡酰奎寧酸,4, 5-O-二咖啡酰奎寧酸和黃芩苷的平均回收率分別為98.62 %,102.58 %,100.41 %,104.76 %,103.17 %,102.95 %,97.39 %,RSD分別為1.93 %,1.08 %,1.10 %,0.74 %,0.92 %,0.78 %,1.13 %。表明測定方法的回收率良好。
2.5.4.6 色譜柱耐用性考察 分別考察色譜柱2~20、22、24~25,各成分分離度良好,且各成分與綠原酸的f值的色譜柱間RSD均小于2.0 %。
2.5.5 樣品含量測定 精密吸取供試品溶液10 μl,注入液相色譜儀,平行測定2次,分別采用雙標多測法和外標法測定樣品中7個成分的含量,結果見表4。比較兩種方法測得含量,絕對偏差(AD)均小于0.1 mg/ml,表明雙標多測法可用于銀黃口服液中7個成分的含量測定。
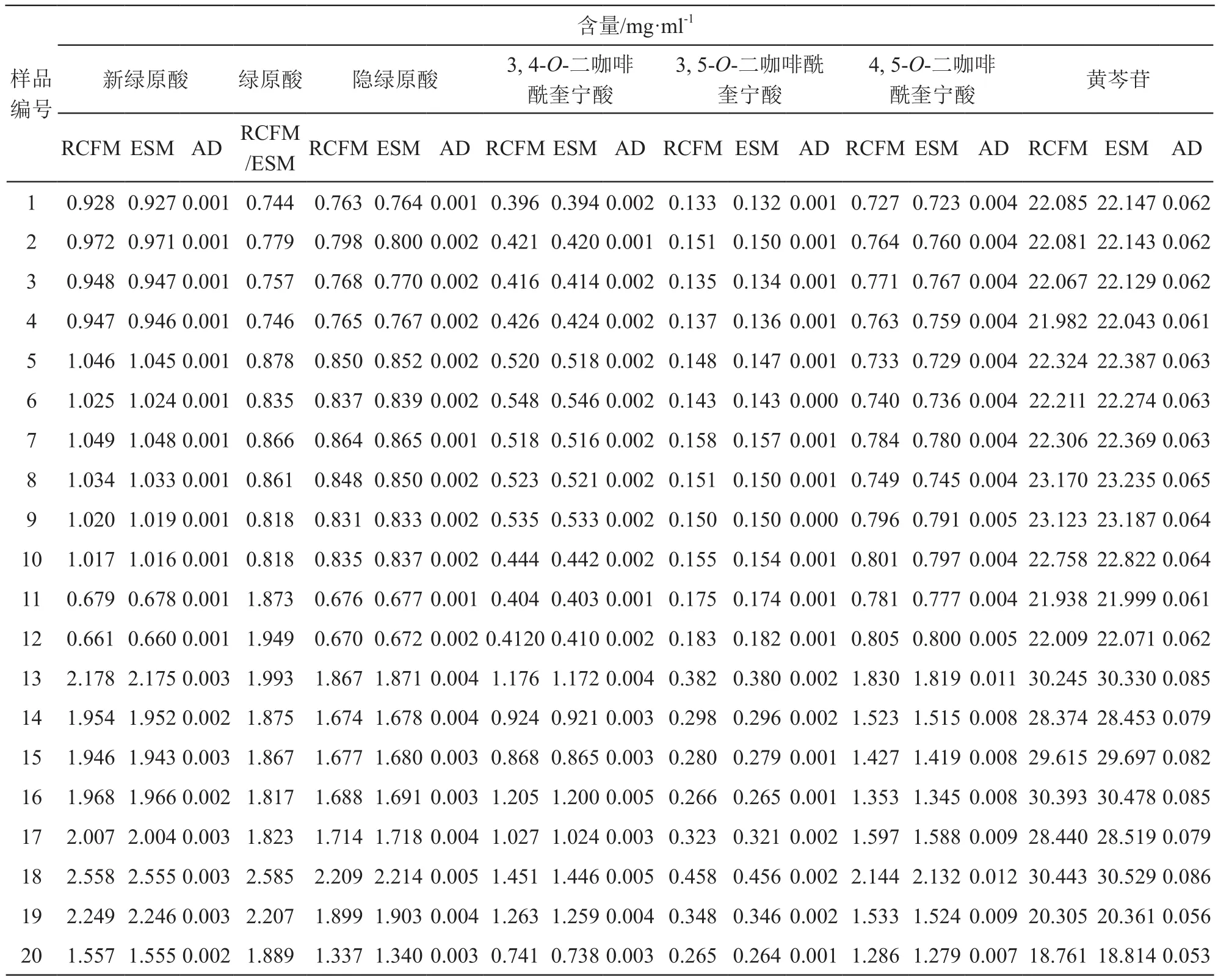
表4 樣品中7個成分含量測定結果(n=2)
3 討論
本文建立了雙標多測法測定銀黃口服液中6種咖啡酰奎寧酸和黃芩苷的含量,以綠原酸和黃芩苷為雙標化合物,采用雙標線性校正法進行色譜峰定性,同時以雙標化合物中的綠原酸作為新綠原酸、隱綠原酸、3, 4-O-二咖啡酰奎寧酸、3,5-O-二咖啡酰奎寧酸、4, 5-O-二咖啡酰奎寧酸和黃芩苷的替代對照品,采用最大吸收波長相對校正因子法對銀黃口服液進行定量研究。有研究表明,檢測波長和儀器波長偏移對相對校正因子影響較大[7],測定相對校正因子時應采用各組分最大吸收波長模式而非單一波長模式,前者能避免因儀器和環境因素改變引起的誤差。將雙標多測法與外標法進行對比,兩種方法所測得結果絕對偏差均小于0.1 mg/ml。
中藥品種眾多且成分復雜,現行標準中含量測定項大多僅控制單一成分,因此替代對照品法有著廣闊的應用前景。雙標多測法是對替代對照品法的一種發展,提供了一種替代對照品法研究的新思路,這一方法將大大降低中藥及天然藥物分析檢測的成本。但作為一種新的應用方法,在標準化和實用化方面,尚需進一步研究和驗證。
