混合型反相微乳體系萃取茶渣蛋白的工藝優化
2022-08-03 15:24:26譚梓銘解新安
食品工業科技 2022年14期
關鍵詞:體系
張 陽,代 成,譚梓銘,李 璐,李 雁,解新安,2,
(1.華南農業大學食品學院,廣東廣州 510642;2.廣東省功能食品活性物重點實驗室,廣東廣州 510642)
茶葉是一種起源于中國的產品,是一種天然的健康飲料,在世界各地工廠的加工過程中,會產生大量的茶葉殘渣[1-2]。據報道,茶渣中含有多種活性成分,其中粗蛋白含量為20%~30%,且80%以上為非水溶性蛋白質[3]。茶渣蛋白由多種氨基酸組成,具有較高的營養價值,此外,它還具有較好的防輻射[4]、抗氧化[5]和降血脂的功能[6],是一種有著極大開發潛力的優質植物蛋白源[7]。目前,茶渣蛋白主要是通過堿提取法[8]和酶提取法[9]來進行萃取。然而,這些方法有著一定的局限性,如環境污染和資源消耗等問題,無法大規模生產。
微乳提取被認為是取代傳統方法的理想選擇[10-11]。微乳是一個單一的光學各向同性且熱力學穩定的體系,是一種含有水相、油相和表面活性劑的自發形成的宏觀分散溶液[12-13]。其中常見的微乳為水包油(O/W)型,而油包水(W/O)微乳也稱為反相微乳。已經證明了W/O 微乳體系中的“水池”比純水有更好的溶解能力[14]。因此,W/O 型微乳已被廣泛用于植物蛋白的提取[15]。反相微乳萃取茶渣蛋白的過程包括兩個步驟:前萃和后萃,其中蛋白質從水相進入表面活性劑聚合物的水池中稱為前萃,而含有這些蛋白質的溶液從反膠束被回收到新的水溶液中的過程稱之為后萃[16-17]。
已有報道表明,反相微乳提取的植物蛋白具有更好的功能特性和結構[18]。Zhao 等[19]使用AOT 反膠束提取大豆蛋白,發現其具有更好的功能、營養和風味特性,并且FTIR 和氨基酸分析結果表明,大豆蛋白在AOT 反膠束中幾乎保持了原有的結構。可能是因為反相微乳中的反膠束結構對植物蛋白有著一定的保護作用[20-21]。此外,Zhu 等[22]研究發現,與堿提法和等電點沉淀法制備的蛋白相比,反膠束法提取的脫脂小麥胚芽蛋白具有相對較好的氮溶性、脂肪吸收能力、起泡性、起泡穩定性和乳化穩定性,結構也更加致密有序。因此使用反相微乳能夠從茶渣中提取優質的茶渣蛋白,但目前用反相微乳法從茶葉殘渣中提取蛋白質的研究鮮有報道。
在本實驗室以前的研究中,Tween80、AOT、SDS和CTAB 四種表面活性劑制成的反相微乳對茶渣蛋白的提取效率不高,因此本文使用Tween80、AOT、SDS 和CTAB 制備了Tween80-AOT、Tween80-CTAB、Tween80-SDS 三種反相微乳體系,并研究了各種因素(前萃:表面活性劑濃度、離子型表面活性劑含量、pH、W0、萃取溫度和萃取時間,后萃取:KCl 濃度、緩沖液pH 和提取溫度)對反相微乳提取茶渣蛋白的影響。然后通過正交試驗對提取條件進行了優化,并通過SDS-PAGE 電泳測定了由反相微乳提取的茶渣蛋白的分子量,為后續生產中進一步開發深加工的茶渣資源提供理論依據。
1 材料與方法
1.1 材料與儀器
茶葉 廣州曼陀嶺茶廠提供;AOT、SDS、CTAB、KCl 溶液 廣州裕兆科技有限公司提供;Tween80、異辛烷、正辛醇 廣州光華科技有限公司;一級水實驗室自制;所有有機溶劑 均為分析純。
PL203 電子天平 梅特勒-托利多儀器有限公司;DF-15 型中藥粉碎機 溫嶺市大德中藥機械有限公司;KQ-100 超聲波清洗機 昆山市超聲儀器有限公司;磁力攪拌器 江蘇市富華有限公司;HH-2 數顯恒溫水浴鍋 常州澳華儀器有限公司;KJELTEC 8200 快速定氮儀 福斯特卡托公司;DL-5 高速離心機 上海安亭科學儀器廠;FD-EC-80 冷凍干燥機、PHS-3C 數顯pH 計 上海精科儀器廠。
1.2 實驗方法
1.2.1 茶渣的制備 將蒸餾水加熱至沸騰狀態,然后將茶葉按料液比1:40 加入并煮沸提取20 min。隨后,用棉布將提取物過濾出來。上述處理重復2次后放入烘箱干燥至恒重,然后用DF-15 型中藥粉碎機將茶渣磨碎。磨碎的茶渣用60 目網篩過篩[23]。
1.2.2 反相微乳的制備 將一定濃度的表面活性劑溶解在由異辛烷和正辛醇組成的有機溶劑中,得到反相微乳,三種混合表面活性劑(Tween80-AOT、Tween80-CTAB、Tween80-SDS)由兩種不同的表面活性劑按一定質量比(w/w)混合組成。然后用磁力攪拌器攪拌混合后的溶液,直到完全溶解。反相微乳體系中的水含量表示為W0(水與表面活性劑的摩爾比),用含有氯化鉀的磷酸鹽緩沖液調節離子強度和pH。然后,溶液在超聲波條件下反應30 min,直到其清晰透明[24]。
1.2.3 蛋白質含量的測定 茶渣蛋白含量的測定參考GB 5009.5-2010《食品安全國家標準食品中蛋白質的測定》中的凱氏定氮法。
1.2.4 蛋白質前萃實驗 取按試驗要求配制的反相微乳置于錐型瓶中,加入一定量的茶渣,超聲一定的時間,然后4000 r/min 離心分離20 min。萃取體系分為兩層,上層為萃取蛋白質的反相微乳層,下層為殘渣,除去殘渣,測量上清液(即為前萃液)體積,取5 mL 上清液定氮(以萃取前的反相微乳作為空白樣)[25]。然后以茶渣蛋白前萃率為指標,考察表面活性劑濃度(0.02、0.04、0.06、0.08、0.10、0.12、0.14 g/mL)、離子型表面活性劑質量分數ω(占表面活性劑總質量的比例,10%、20%、30%、40%、50%、60%、70%、80%、90%)、W0(5、10、15、20、25、30、35)、水相pH(7、8、9、10、11、12、13)、萃取溫度(25、30、35、40、45、50、55、60 ℃)以及萃取時間(20、40、60、80、100、120 min)5 種因素不同水平對前萃過程的影響。固定考察因素表面活性劑濃度0.08 g/mL,離子型表面活性劑質量分數50%,W025,水相pH10,萃取溫度40 ℃,萃取時間60 min。蛋白質前萃率由下式計算:

根據單因素實驗的結果,選擇了表面活性劑濃度(A)、離子型表面活性劑的質量分數(B)、水相增溶能力W0(E)和水相pH 四個因素。以茶渣蛋白前萃率為指標,通過正交試驗確定最佳提取工藝,因素水平設計如表1所示。

表1 前萃正交試驗因素與水平L16(45)Table 1 Factors and levels of forward extraction orthogonal test L16(45)
1.2.5 蛋白質后萃取實驗 取10 mL 前萃液置于錐形瓶中,加入等體積的具有一定離子強度和pH 的KCl 緩沖液,超聲波萃取一定時間,然后4000 r/min離心分離20 min。分液漏斗分層后取下層水相,測量體積,取5 mL 下層水相定氮(以KCl 緩沖液作為空白樣),余者真空冷凍干燥,得粗蛋白,測定蛋白質含量[25]。然后以茶渣蛋白后萃率為指標,考察了緩沖液KCl 濃度(0.25、0.5、1.0、1.5、2.0 mol/L)、緩沖液pH(5.5、6.0、6.5、7.0、8.0、9.0、10.0、11.0、12.0)以及萃取溫度(25、30、35、40、45、50、55、60 ℃)三個因素不同水平對后萃過程的影響。后萃單因素實驗初始條件為:KCl 濃度0.08 mol/L、水相pH7 以及萃取溫度40 ℃。蛋白質后萃率由下式計算:

通過正交試驗進一步研究了茶渣蛋白后萃的最佳條件。如表2所示,對氯化鉀的濃度(A)和pH(B,C)以及提取溫度(D)進行考察,每個因素選取三個水平,進行正交優化試驗。
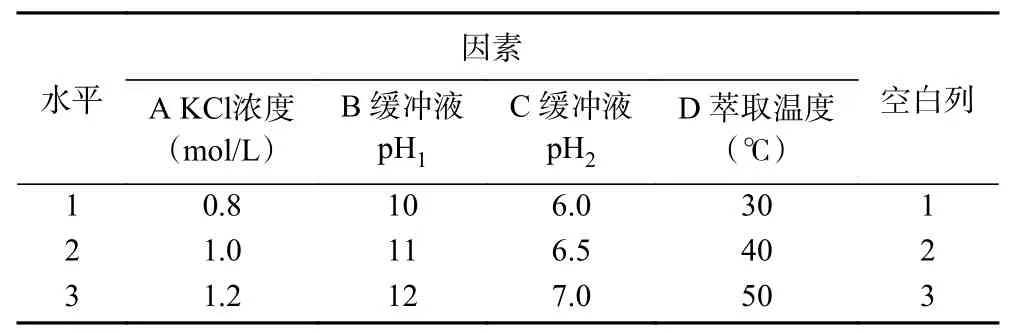
表2 后萃正交試驗因素與水平L9(34)Table 2 Factors and levels of forward extraction orthogonal testL9(34)
1.2.6 茶渣蛋白分子量測定 參照何忠效的《生物化學試驗技術》中的試驗方法。使用4%的濃縮膠和15%的分離凝膠進行SDS-PAGE 電泳分析。含有相同數量的蛋白質的樣品與分子量的標記蛋白一起被加載到丙烯酰胺凝膠上。電泳步驟結束后,使用考馬斯亮藍R-250 染色1 h,然后用脫色液(乙醇:乙酸:蒸餾水=25:8:67)脫色2 h。
1.3 數據處理
實驗結果為重復三次實驗后,取平均值。所有的實驗數據都使用SPSS 軟件包(11.5 版)進行分析,并使用Excel 2013(Microsoft Corporation,Redmond,WA,USA)繪制了曲線。然后進行方差分析以評估獨立之間的顯著差異,并通過鄧肯多重范圍檢驗(DMRT)評估平均值之間的差異是否顯著,不同的小寫字母表示具有顯著性差異(P<0.05)。
2 結果與分析
2.1 茶渣蛋白前萃實驗結果
2.1.1 表面活性劑濃度對蛋白質前萃率的影響 由圖1可知,隨著表面活性劑濃度的增加,Tween80-SDS 和Tween80-CTAB 反相微乳的前萃率先增加后減少,當表面活性劑濃度為0.10 g/mL 時,前萃率達到最高值。Tween80-AOT 反相微乳體系的前萃率低于上述兩種微乳體系,當表面活性劑濃度為0.08 g/mL 時,萃取率達到最大值9.5%。這一結果可能是表面活性劑濃度的影響造成的,表面活性劑的濃度升高后增強了微乳中水的增溶作用,從而增大了膠束的尺寸和/或增加了膠束的數量[26],使蛋白質更有選擇性地增加轉移到有機相。另一方面,表面活性劑濃度過高,不利于茶渣蛋白的前萃取。因此選擇0.08 g/mL 的表面活性劑濃度(Tween80-AOT)和0.10 g/mL 的 表 面 活 性 劑 濃 度(Tween80-SDS 和Tween80-CTAB)進行后續實驗。
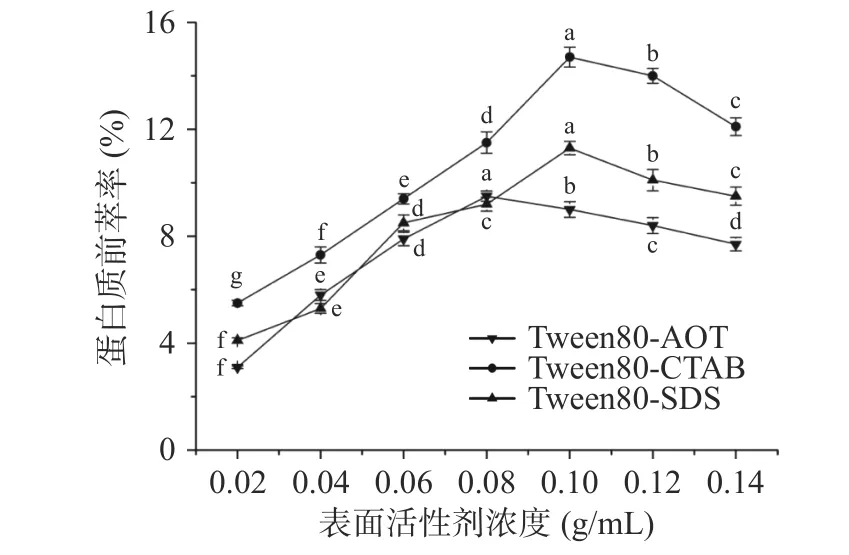
圖1 表面活性劑濃度對前萃率的影響Fig.1 Effect of surfactant concentration on the forward extraction
2.1.2 離子型表面活性劑質量分數對蛋白質前萃率的影響 在離子型表面活性劑含量增加的條件下(圖2),三種不同的反相微乳體系提取的茶渣蛋白含量均迅速攀升,然后下降。在Tween80-CTAB 和Tween80-SDS 微乳體系中,CTAB 和SDS 含量為70%時,萃取率達到最高,分別為15.1%和11.9%。而在Tween80-AOT 微乳體系中,AOT 含量從10%到80%變化時,前萃率逐漸增加,且AOT 含量增加到80%時,茶渣蛋白的前萃率接近最大值(10.9%),然而,當AOT 含量進一步增加時,前萃率反而開始下降。因此在配制混合反相微乳時,本實驗選取AOT 的含量為80%,選取CTAB 與SDS 的含量為70%。
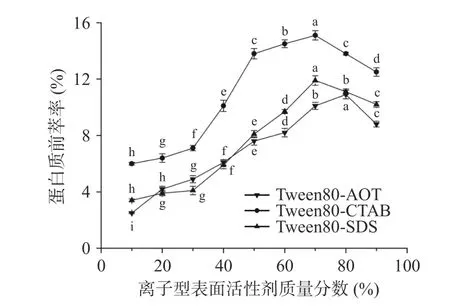
圖2 離子型表面活性劑質量分數對前萃率的影響Fig.2 Effect of ionic surfactant content on the forward extraction
2.1.3 W0對蛋白質前萃率的影響 當W0從5 增加到30 時,Tween80-CTAB、Tween80-AOT 和Tween80-SDS 反相微乳體系的前萃率都有極大的提升(圖3),當W0達到25 時,前萃率的增加趨勢趨于平緩。W0的增加對應于膠束大小的增加,這與蛋白質的溶解度密切相關。這些現象可能是因為隨著W0的增加,一些較大的反相膠束形成,可以包含多個蛋白質分子[27]。因此,最佳的W0應該是25。
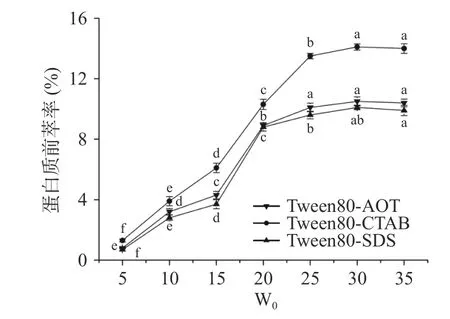
圖3 W0 對前萃率的影響Fig.3 Effect of W0 on the forward extraction
2.1.4 水相pH 對蛋白質前萃率的影響 從圖4 可以看出,在三種反相微乳體系中中,茶渣蛋白前萃率的變化與pH 的變化趨勢不同。對于Tween80-AOT和Tween80-SDS 微乳體系,前萃率先上升然后略有下降,當pH 為9 時達到最大值。對于Tween80-CTAB微乳體系,前萃率隨著pH 的增加而增加,但當pH達到12 時,茶渣蛋白的前萃率(14.2%)最高。相比于其他兩種反相微乳體系在相同pH 下的提取效果,Tween80-CTAB 微乳體系對茶蛋白的提取率最高。在最佳pH 下,蛋白質的提取率會通過靜電相互作用或疏水相互作用得到改善[16]。過高或過低的pH 不僅會導致蛋白質變性,而且由于將蛋白質和表面活性劑混合成乳狀液而無法形成反相膠束[28]。后續實驗Tween80-AOT 和Tween80-SDS 微乳體系的水相pH選擇為9.0,而Tween80-CTAB 微乳體系的水相pH選擇12.0。
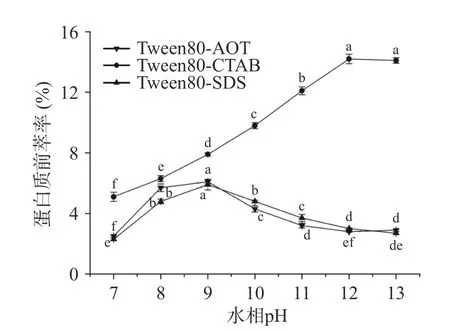
圖4 pH 對前萃率的影響Fig.4 Effect of pH on the forward extraction
2.1.5 萃取溫度對蛋白質前萃率的影響 如圖5所示,隨著溫度的升高,微乳的整體萃取趨勢表現為先升高然后降低。當萃取溫度為45 ℃時,Tween80-AOT 反相微乳體系獲得了最大的萃取率,而當溫度為40 ℃時,Tween80-CTAB 體系和Tween80-SDS體系的萃取率最高,然后當溫度繼續上升時,萃取率逐漸開始下降。這表明溫度過高或過低都不利于蛋白質的前萃取,不恰當的溫度會影響蛋白質在微乳中的溶解度[29-30]。而在適當的溫度下,蛋白質和反相膠束分子之間的相互作用會增強[31]。因此,本實驗中Tween80-AOT 微乳體系的適宜萃取溫度為45 ℃,Tween80-SDS 和Tween80-CTAB 微乳體系的適宜提取溫度為40 ℃。
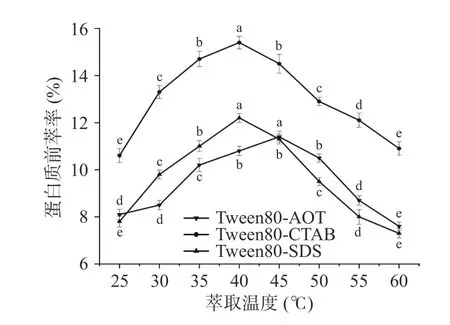
圖5 萃取溫度對前萃率的影響Fig.5 Effect of temperature on the forward extraction
2.1.6 萃取時間對蛋白質前萃率的影響 在圖6 中,前萃率在提取時間為20~60 min 的范圍內明顯增加,隨后在60~120 min 之間的上升趨勢趨于平緩。這表明在早期階段(20~60 min),蛋白質的前萃取容易受到提取時間的影響,而且提取率隨著提取時間的增加而上升。在60~80 min 之間,茶渣蛋白的前萃率沒有隨著提取時間的增加而發生明顯的改變,這可能是由于在前期的萃取過程中,反膠束已經增溶了大量的茶渣蛋白,達到一個飽和狀態,并且隨著萃取時間的增加,離子強度等因素也會對反膠束結構造成一定的影響[31],因此在隨后實驗中萃取時間選擇40 min。
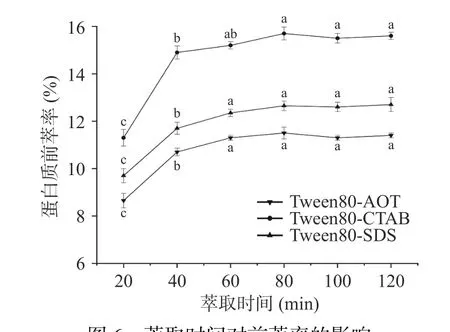
圖6 萃取時間對前萃率的影響Fig.6 Effect of extraction time on the forward extraction
2.1.7 前萃工藝的正交試驗 根據上述單因素實驗的結果,對影響前萃取的四個主要因素(表面活性劑濃度、離子型表面活性劑的質量分數、W0和水相的pH)進行了正交優化試驗。從表3 中正交試驗結果可以看出,影響Tween80-AOT、Tween80-CTAB 和Tween80-SDS 三種反相微乳體系前萃率的主次順序分別是W0>pH>表面活性劑濃度>離子型表面活性劑質量分數、表面活性劑濃度>pH>W0>離子型表面活性劑質量分數和表面活性劑濃度>pH>離子型表面活性劑質量分數>W0。且從表4~表6 中可以看出,僅有W0對Tween80-SDS 體系的前萃率影響不顯著。實驗結果表明,Tween80-AOT 微乳體系中四個因素的最佳組合是:A3B2C4E3,即在表面活性劑濃度為0.10 mol/L、離子型表面活性劑含量為70%、pH 為10.0、W0為25 的條件下,Tween80-AOT 微乳體系的茶渣蛋白前萃率最高(11.49%)。Tween80-CTAB 微乳中四個因素的最佳組合是:A3B2D4E3,茶蛋白的最大前萃率的條件為:表面活性劑濃度0.10 mol/L,離子型表面活性劑含量70%,pH13.0,W025,最高前萃率為16.17%。Tween80-SDS 體系中四個因素的最佳組合是:A2B3C3E3,在最佳條件下(表面活性劑濃度為0.08 mol/L,SDS 含量為80%,pH 9.0,W025)進行實驗后,前萃率為13.10%。
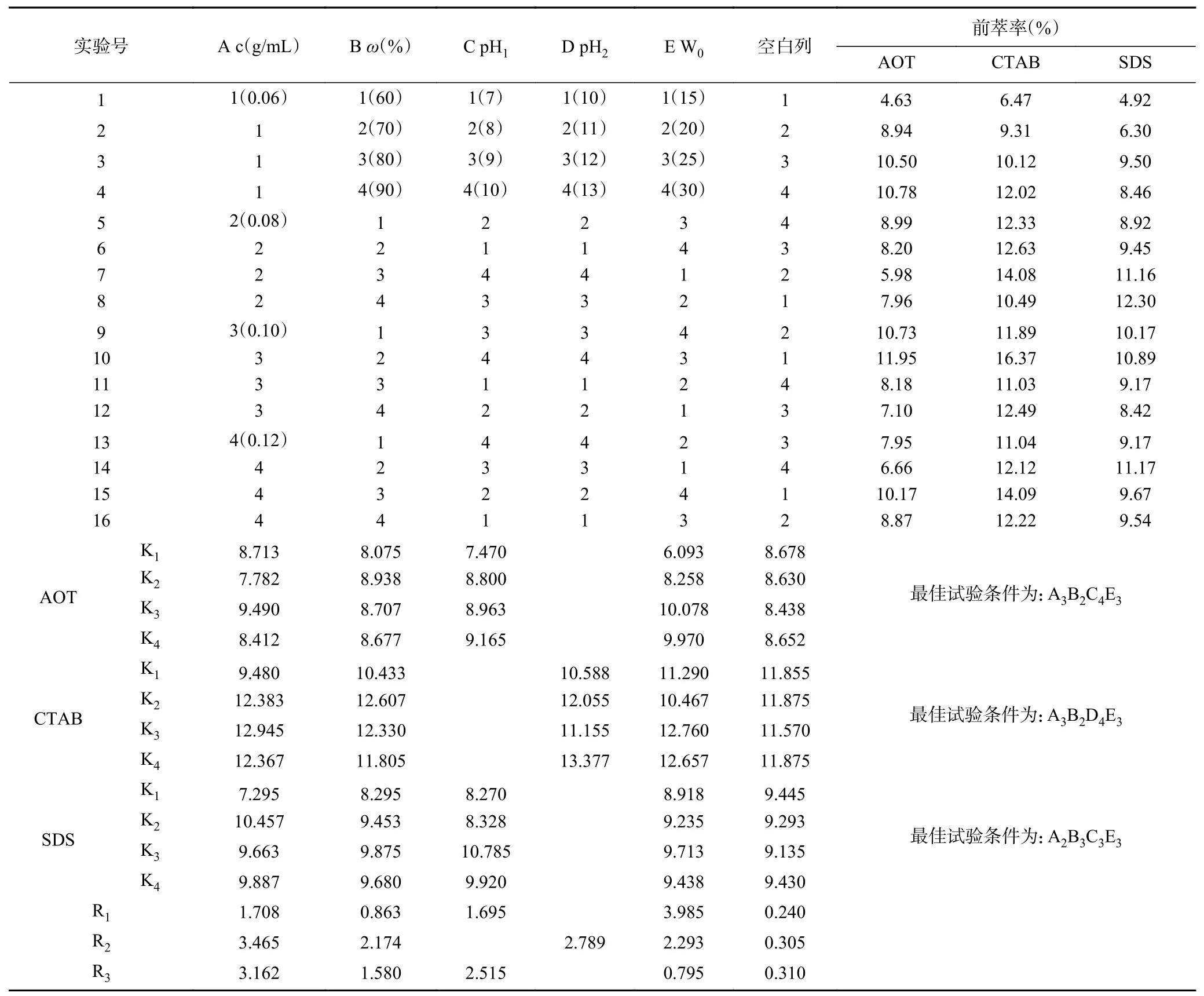
表3 前萃正交試驗安排與結果Table 3 Arrangement and results of forward extraction orthogonal experiment

表4 Tween80-AOT 體系前萃正交試驗方差分析表Table 4 Significant analysis on forward extraction orthogonal test of Tween80-AOT system

表6 Tween80-SDS 體系前萃正交試驗方差分析表Table 6 Significant analysis on forward extraction orthogonal test of Tween80-SDS system

表5 Tween80-CTAB 體系前萃正交試驗方差分析表Table 5 Significant analysis on forward extraction orthogonal test of Tween80-CTAB system
2.2 茶渣蛋白后萃實驗結果
2.2.1 緩沖液KCl 濃度對蛋白質后萃率的影響 圖7顯示了KCl 濃度對后萃率的影響。隨著KCl 濃度的增加,三種反相微乳體系的后萃率均表現為先上升后下降,當KCl 濃度為1.0 mol/L 時達到最大值,其中Tween80-CTAB 最大后萃率為94.5%,而Tween80-AOT 和Tween80-SDS 最大后萃率分別為90.21%和89.41%。研究發現,隨著KCl 濃度的增加,表面活性劑周圍的電層厚度逐漸變薄,表面活性劑的極性頭之間的相互排斥力降低,導致反相微乳中的膠束變小,使得微乳中蛋白質的增溶作用減弱,后萃率得到提高[32]。
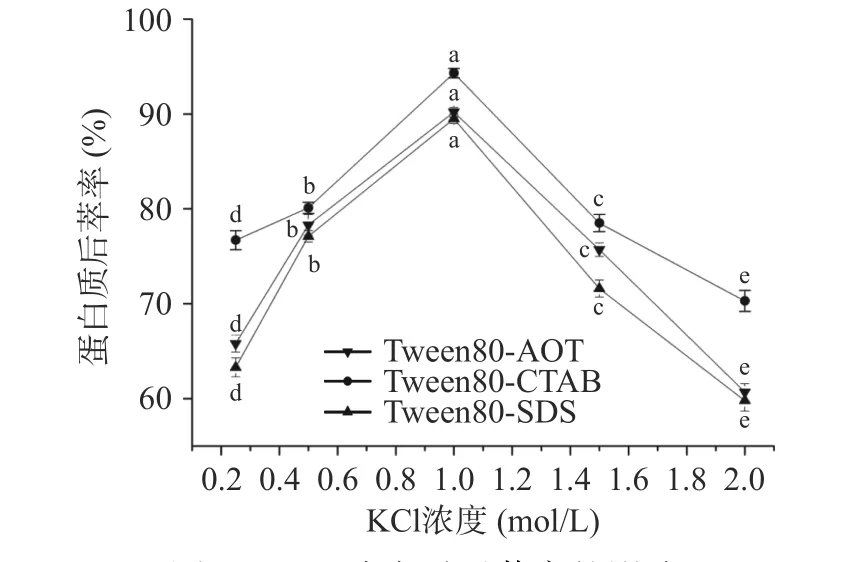
圖7 KCl 濃度對后萃率的影響Fig.7 Effect of KCl concentration on the backward extraction
2.2.2 緩沖液pH 對蛋白質后萃率的影響 如圖8所示,Tween80-AOT 和Tween80-SDS 微乳體系的茶渣蛋白后萃率隨著pH 的升高而增大,當pH 在5.5~11 時,茶渣蛋白的后萃率明顯上升,在pH 到達11 后,出現緩慢下降的趨勢,因此在pH 為11 時達到最高值。在Tween80-CTAB 體系中,pH 在5.5~6.5時,蛋白質的后萃率大大增加,然后隨著pH 的增加逐漸降低,茶渣蛋白的后萃率在pH 6.5 時達到最高。緩沖液pH 影響后萃率的原因是隨著pH 的增加,更多的茶渣蛋白分子帶負電,電荷密度增加,削弱了蛋白質與表面活性劑之間的靜電作用,促進了后萃取過程,即蛋白質從反相膠束中的釋放率增加[33]。另一方面,較高的pH 反而會增大蛋白質之間的靜電排斥作用,導致蛋白質后萃率下降[34]。

圖8 緩沖液pH 對后萃率的影響Fig.8 Effect of buffer pH on the backward extraction
2.2.3 萃取溫度對蛋白質后萃率的影響 反相微乳中茶蛋白的提取率可能受到溫度的影響[24]。從圖9中可以得知,三種反相微乳體系對茶渣蛋白的提取率趨勢一致,且在40 ℃時后萃率達到最高值,但隨著溫度的持續升高,后萃率均明顯下降。原因是在溫度升高之初,反相微乳與水相之間的分子運動加快,反相微乳的膠束破壞變大,蛋白質的后萃率增加。當溫度繼續升高,萃取率反而開始下降,且過高的溫度會導致蛋白變性[25]。因此,三種反相微乳體系的最佳萃取溫度為40 ℃。
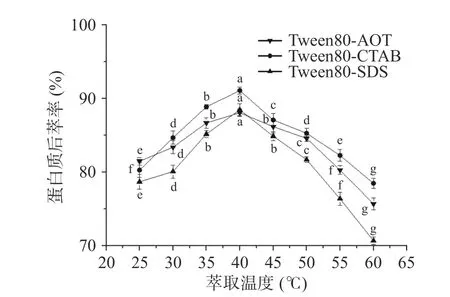
圖9 萃取溫度對后萃率的影響Fig.9 Effect of temperature on the backward extraction
2.2.4 后萃正交試驗 從表7 中正交試驗結果可以看出,影響Tween80-AOT、Tween80-CTAB 和Tween80-SDS 三種反相微乳體系后萃率的主次順序分別是萃取溫度>KCl 濃度>緩沖液pH、KCl 濃度>緩沖液pH>萃取溫度和萃取溫度>KCl 濃度>緩沖液pH。且從表8~表10 中可以看出,三個因素對三種反相微乳體系都有顯著影響。實驗結果表明,在KCl 濃度為1.2 mol/L、pH 為11.0、萃取溫度為50 ℃的條件下,茶渣蛋白在Tween80-AOT 微乳體系中的后萃率最高(91.53%),則三個因素的最佳組合是:A3B2D3。Tween80-CTAB 微乳體系中的三個因素的最佳組合是:A3C3D2,即KCl 濃度為1.2 mol/L,pH 為7.0,萃取溫度為40 ℃,達到最高后萃率94.78%。表中觀察到Tween80-SDS 微乳體系中茶渣蛋白的最佳后萃取條件為KCl 濃度1.2 mol/L,pH11.0,提取溫度30 ℃,則三個因素的最佳組合是:A3B2D1,在最佳條件下茶渣蛋白的后萃率為87.72%。
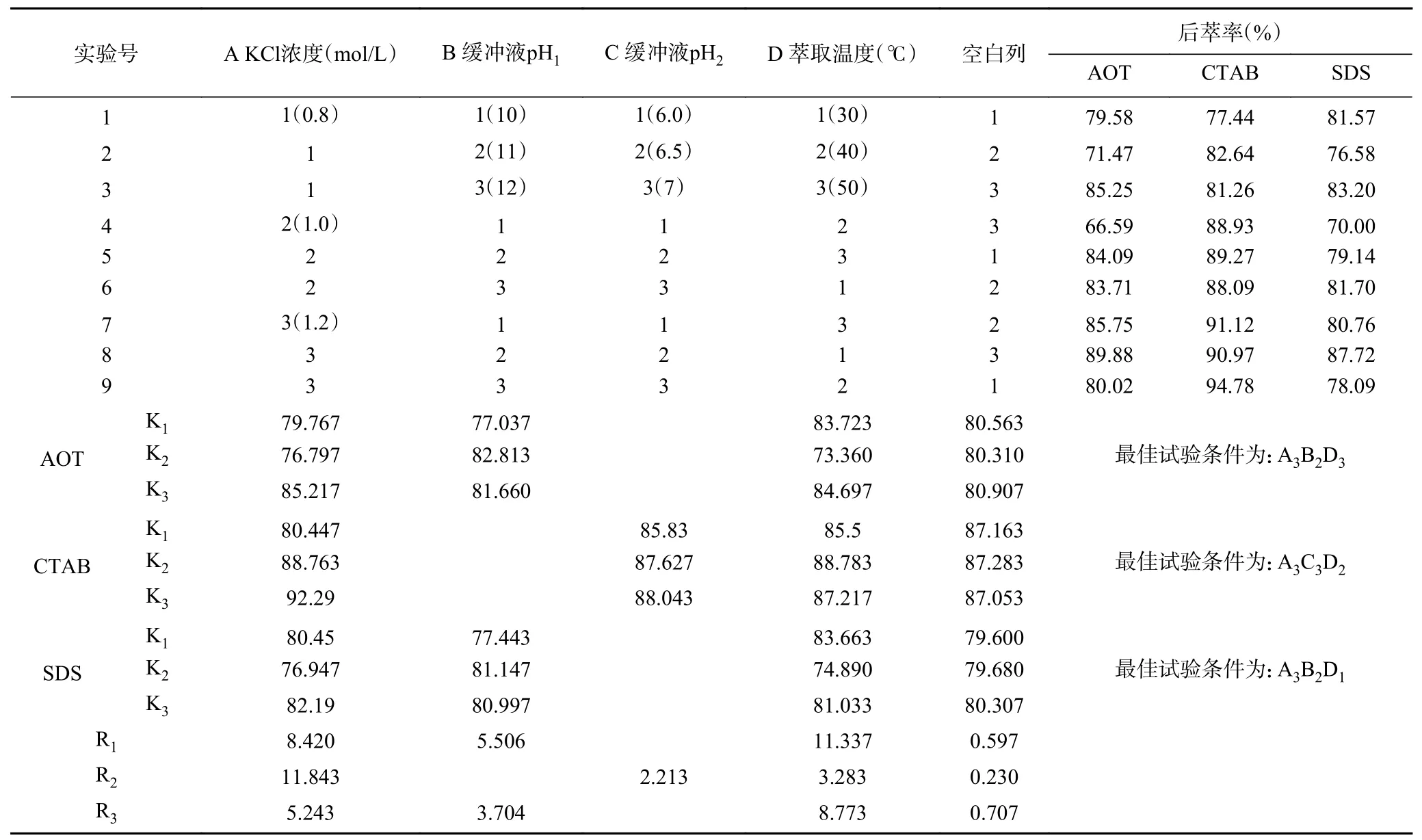
表7 后萃正交試驗安排與結果Table 7 Arrangement and results of backward extraction orthogonal experiment
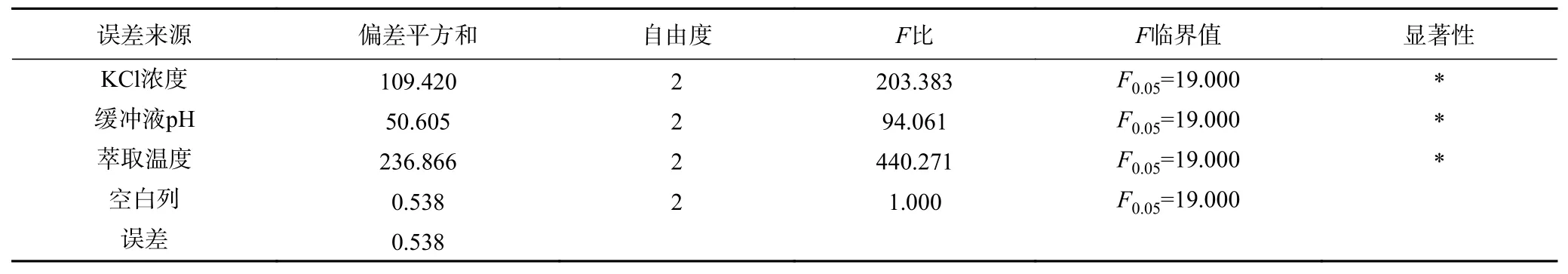
表8 Tween80-AOT 體系后萃正交試驗方差分析結果Table 8 Significant analysis on backward extraction orthogonal test of Tween80-AOT system
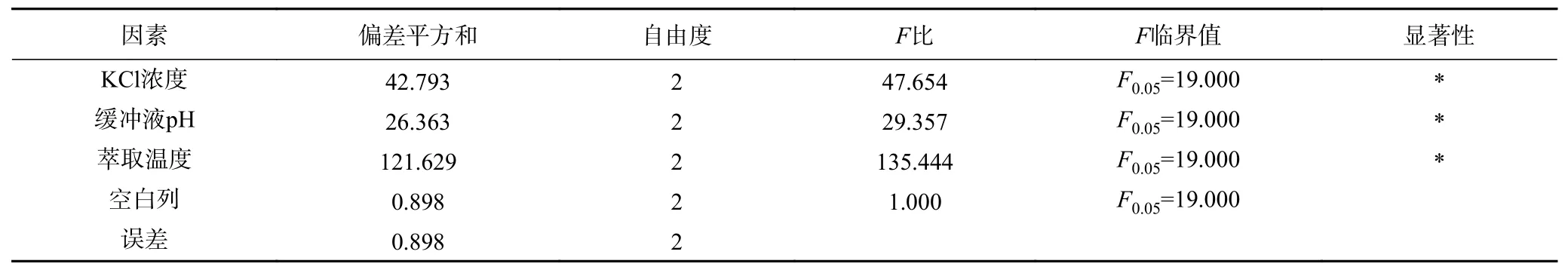
表10 Tween80-SDS 體系后萃正交試驗方差分析結果Table 10 Significant analysis on backward extraction orthogonal test of Tween80-SDS system
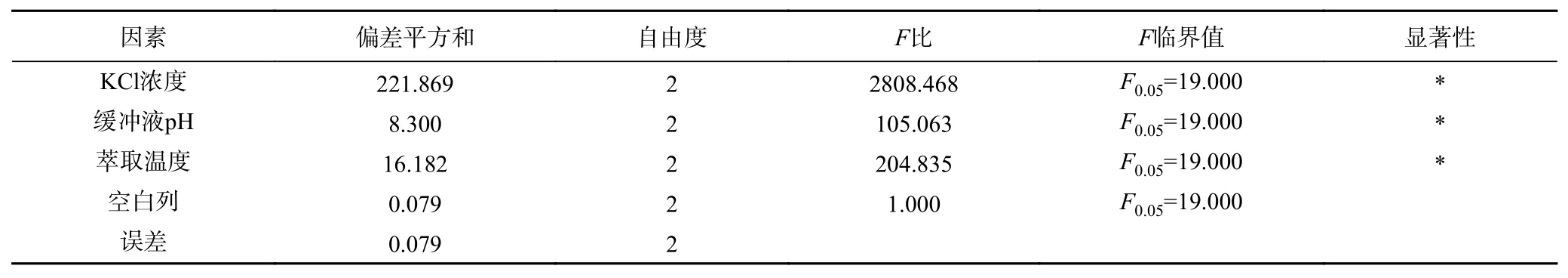
表9 Tween80-CTAB 體系后萃正交試驗方差分析結果Table 9 Significant analysis on backward extraction orthogonal test of Tween80-CTAB system
2.3 茶蛋白分子量測定結果
圖10展示了用Tween80-CTAB 反相微乳提取茶渣蛋白的SDS-PAGE 電泳圖。在茶渣蛋白的光譜中,有兩條帶子,兩條帶子的分子量分別為27.18 和20.85 kDa,小于堿法提取茶渣蛋白的分子量。在以前的工作中,我們研究了不同方法(反相微乳法、堿法和酶法)提取的茶渣蛋白的特性,結論表明,反相微乳法提取的茶葉蛋白與堿法和酶法提取的蛋白相比,具有更好的溶解性和乳化性等功能特性[23]。根據SDS-PAGE 電泳的結果可知,堿溶法提取的茶渣蛋白的分子帶多于反轉微乳法提取的茶葉蛋白。因此,可以知道反相微乳可以選擇性地提取高活性的茶渣蛋白。
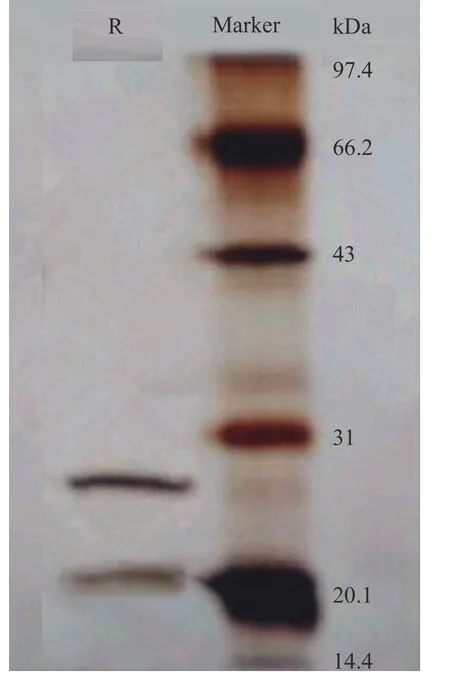
圖10 茶渣蛋白分子量Fig.10 Molecular weight of tea residues protein
3 結論
本研究用非離子型表面活性劑(Tween80)和離子型表面活性劑(AOT、SDS、CTAB)制備了3 種混合表面活性劑的反相微乳體系用于提取茶渣蛋白。實驗結果表明,從茶葉殘渣中提取高活性的茶葉蛋白,Tween80-CTAB 反相微乳是最佳選擇,其最佳提取條件為表面活性劑濃度0.10 mol/L,離子型表面活性劑含量70%,pH13.0,W025,萃取溫度40 ℃,萃取時間40 min。在這種條件下,茶渣蛋白的前萃率為16.17%。然后在最佳條件(氯化鉀濃度為1.2 mol/L,pH7.0,提取溫度40 ℃)下進行后萃實驗后,得到其最佳后萃率為94.78%。這些數據證明了用反相微乳法從茶葉殘渣中提取蛋白質的可行性,為如今大量存在的廢棄茶渣提供了一個切實可行的處理方法,同時也減少了廢棄茶渣對環境的污染。另一方面,未來的研究還需要進一步提高茶渣蛋白的前萃率,使茶渣能夠得到更充分的利用。SDS-PAGE 結果顯示,反相微乳可以選擇性地提取分子量較小的高活性茶渣蛋白,這可能是前萃率較低的原因。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
