基于動態網絡切分的關鍵蛋白質預測方法
2022-07-12 02:45:04鐘堅成瞿佐航
計算機研究與發展 2022年7期
鐘堅成 方 卓 瞿佐航 鐘 穎 彭 瑋 潘 毅
1(湖南師范大學信息科學與工程學院 長沙 410081) 2(昆明理工大學信息工程與自動化學院 昆明 650500) 3(中國科學院深圳理工大學計算機與控制工程學院 廣東深圳 518055)
蛋白質作為構成一切生物細胞和結構必不可少的組成部分,與所有生物的生命活動息息相關,是生理功能的“基石”.由于不同氨基酸的排列順序和空間組合有所差異,導致了蛋白質在各種生命活動中扮演了不同的角色.蛋白質可區分為關鍵蛋白質和非關鍵蛋白質,關鍵蛋白質在生物體內的作用尤為重要,正常生物體內如果缺少某類關鍵蛋白質,會造成生物體內某項功能的喪失,影響其正常生命活動,進而會導致該生物體的死亡[1].因此,準確有效地識別關鍵蛋白質對于研究疾病的源頭和生物細胞的生長調控過程具有重要意義.
1 相關工作
基于生物實驗的關鍵蛋白質驗證方法有較高的準確性,但存在實驗周期較長和消耗大量人力、財力等局限.針對此問題一些基于計算的方法被研究學者提出,分為有監督和無監督方法.有監督方法是訓練一個預測模型從已標記的樣本中學習特征來訓練模型以預測關鍵蛋白質.一些經典的機器學習方法包括邏輯回歸、隨機森林、決策樹、支持向量機(SVM)、神經網絡等都屬于有監督的方法.Hwang等人[2]將不同種類的蛋白質相互作用(protein-protein interaction, PPI)網絡拓撲特征,并結合開放閱讀框(open reading frame, ORF)長度、種系保留(PHY)等生物特征,利用SVM方法對關鍵蛋白質進行預測.Acencio等人[3]結合了亞細胞定位的局部效應、生物特征和網絡拓撲特征,利用一種多決策樹投票策略進行預測.Deng等人[4]結合了樸素貝葉斯分類器、C4.5決策樹、CN2規則和邏輯回歸模型預測關鍵蛋白質.Zhong等人[5]結合拓撲特征和生物信息特征,提出了一種XGBFEMF框架預測關鍵蛋白質.Zeng等人[6]提出了一種深度學習框架來整合PPI拓撲特征以及基因表達數據,并利用采樣來解決訓練數據不平衡問題.Peng等人[7]結合隨機游走、神經網絡和SVM對人類關鍵基因進行識別.Dai等人[8]提出了一種多集成方法,將多個基分類器進行結合從而達到提升識別率的目的.
無監督的方法無需訓練模型,主要通過挖掘關鍵蛋白質的特征對蛋白質進行關鍵性打分.基于PPI網絡拓撲特征的經典算法是利用蛋白質相互作用網絡中的拓撲特征給蛋白質打分.如:節點的度中心性(degree centrality, DC),根據蛋白質節點在網絡中節點的度的大小來衡量節點的重要性[9];節點的介數中心性(betweenness centrality, BC),指某節點出現在其他節點之間的最短路徑的個數[10];節點的子圖中心性(subgraph centrality, SC),通過計算節點在網絡之中所參與的閉合回路的個數來體現節點的重要性[11];節點的特征向量中心性(eigenvector centrality, EC),是利用在網絡鄰接矩陣的主向量中每個頂點的分量來衡量節點的重要性[12];節點的信息中心性(information centrality, IC),通過調和平均路徑數衡量其重要性[13];鄰域中心性(neighborhood centrality, NC),利用相連邊的重要性[14];局部平均連通性的方法(local average connectivity-based method, LAC),利用鄰居節點的平均連通性來衡量節點重要性[15].然而,盡管這些中心性的方法取得了一定的效果,但也存在自身的局限性,網絡中存在的假陽性及假陰性數據降低了網絡的可靠性,對高度依賴網絡結構的中心性方法造成了干擾.為了降低PPI網絡中假陽性和假陰性對實驗造成的影響,一些研究人員通過融合蛋白質網絡拓撲特征和蛋白質生物信息來解決假陽性對PPI網絡的影響.Li等人[16]和Tang等人[17]分別提出了新的融合性方法PeC和WDC,通過在PPI網絡的基礎上融合了基因表達數據來提高關鍵蛋白質的識別率.Lei等人[18]結合了網絡拓撲特征、基因表達、基因本體(gene ontology, GO)注釋數據、亞細胞定位和蛋白質復合物,并利用隨機游走算法來對蛋白質進行關鍵性打分.胡健等人[19]融合基因表達、結構域和蛋白質復合物等生物信息構建時序加權網絡識別關鍵蛋白質.Chen等人[20]構建了一種蛋白質-結構域網絡,并基于PageRank算法來推斷關鍵蛋白.Liu等人[21]從統計假設檢驗的角度出發,提出了一種基于p值的中心性計算方法.
此外,基因的表達呈現動態性,而靜態PPI網絡忽略了動態性,無法動態刻畫網絡中蛋白質的相互作用,一些學者融合了基因表達的時序數據,根據基因動態表達的特性構建蛋白質動態關聯網絡,以刻畫不同時刻下的蛋白質相互作用關系[22].例如:Lichtenberg等人[23]通過結合不同時間點的基因表達數據和蛋白質相互作用數據構建了時間序列動態網絡.Xiao等人[24]在靜態PPI網絡的基礎上提出了一種時間序列模型并利用k_sigma原理去除噪聲數據,構建NF-PIN動態網絡.Li等人[25]結合基因表達譜和亞細胞定位信息構建了TS-PIN動態網絡來預測關鍵蛋白質.Li等人[26]在PPI網絡中融合了正交數據,并利用擴展Pareto模型預測關鍵蛋白質.
動態網絡利用了基因表達的動態性進一步完善了網絡,但并未考慮基因的周期性表達的特性.一些學者研究表明,基因在不同周期下的表達存在差異,且在不同表達周期下呈現節律性變化.Spellman等人[27]在釀酒酵母中鑒定了800個滿足細胞周期調節的基因.Rustici等人[28]使用DNA微陣列檢測了分裂酵母的基因的周期性表達對整個細胞周期的控制.Luan等人[29]提出了一種統計框架,利用基因表達數據和周期性表達的引導基因來識別周期性表達基因.為了更進一步提升網絡的可靠性、降低網絡中假陰性及假陽性數據的影響,本文在基因表達動態性的基礎上引入周期性表達的概念,提出了一種動態網絡切分的方法.由于關鍵蛋白質往往在生物體中參與了更多重要的生命活動,表現出更多的“活性”狀態,本文通過構建基因“活性”表達矩陣來對基因表達數據中的噪聲數據進行過濾,將各時刻的表達分類成“活性”與“非活性”表達的狀態.并根據基因“活性”表達矩陣來劃分周期從而刻畫連續時間段內的基因表達的動態變化,有利于從局部衡量蛋白質的“活性”程度,更契合基因表達隨周期的改變而發生變化的特性,從而進一步降低網絡中假陽性與假陰性的影響,提高關鍵蛋白質識別的準確性.
2 基于動態網絡切分的關鍵蛋白質預測方法
2.1 構建基因“活性”表達矩陣
由于基因表達數據是由微陣列或新一代測序技術產生的數據,這類高通量的數據存在著不可避免的噪聲數據,以基因隨時間動態表達的特性為前提,利用在不同時刻下基因所呈現“活性”和“非活性”的性質來去除基因表達數據中噪聲的影響.設置的動態閾值計算公式為:
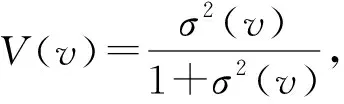
(1)
S(v)=U(v)+a×σ(v)×V(v),
(2)
其中,σ(v)表示基因在整個周期內表達值的標準差,V(v)表示蛋白質基因表達的波動性,U(v)表示基因整個周期的平均表達值,S(v)表示基因的閾值參數,a表示閾值系數.
根據每個基因的表達曲線來設置閾值,如果某個時刻下基因的表達值不高于其閾值,那么該基因的該時刻被認為是“非活性”的表達時刻,對于“非活性”時刻的表達值采用對其定義為“0”,對于高于閾值的“活性”的表達值時刻則維持其原有時刻下的表達值,以此進一步降低其高通量數據帶來的假陽性及假陰性.由此構建的表達值矩陣為:
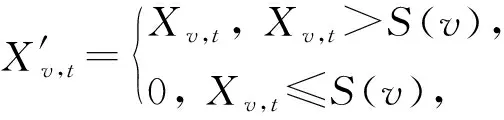
(3)

(4)
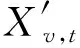
2.2 切分動態網絡子網
蛋白質與蛋白質之間并不總是時刻存在相互作用關系,蛋白質之間的相互作用關系會隨著時間的改變而發生改變.時間序列的基因表達數據為構建動態網絡提供了基礎,在蛋白質的活性動力學基礎上結合基因具有周期表達性的特點,對基因活性表達矩陣進行周期切分.具體切分公式為:
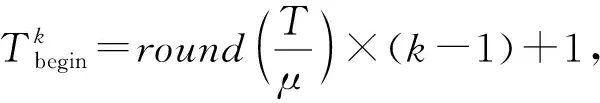
(5)
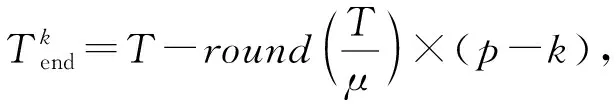
(6)
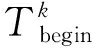
為了反映基因隨周期表達這一特性,動態網絡下2個蛋白質在同一周期下的相同時刻同時存在著“活性”表達,那么此相互作用關系則將在周期網絡中保留.動態網絡切分表示為DDGS={G1,G2,…,Gk,…,Gp},Gk代表第k個周期下的子網,Vk={vk1,vk2,…,vkn}代表第k個周期下的“活性”蛋白質集合,Ek={ek1,ek2,…,ekm}代表第k個周期下基于活性共表達原則的蛋白質相互作用關系集合.對于靜態PPI網絡下的蛋白質相互作用e(v,u),如果蛋白質v和蛋白質u在第k個周期中的時刻t下同時處于“活性”表達,構建的蛋白質相互作用關系集合為:
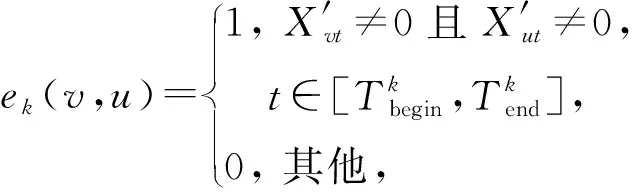
(7)
其中,ek(v,u)=1代表蛋白質v和蛋白質u在第k個周期下存在相互作用關系,反之則不存在相互作用關系.
以p=5,μ=10為例,將酵母(yeast)物種進行動態網絡切分的示意圖如圖1所示.酵母物種的“活性”表達矩陣周期為36個不同時刻,對其進行5個周期的切分,5個周期的起始時間點和終止時間點分別為“活性”表達矩陣的T1與T20,T5與T24,T9與T28,T13與T32和T17與T36,再通過融合靜態PPI網絡相互作用邊來構建子網G1,G2,G3,G4,G5.對于酵母蛋白質周期1的時刻T1至T20來說,A,B存在靜態相互作用,蛋白質A,B在時刻T9下存在“共活性”表達時刻,因此A,B在G1中存在動態子相互作用.B,E即使在時刻T33下存在著“共活性”表達時刻,但在周期1,2,3中不存在“共活性”表達時刻,因此B,E在子網G1,G2,G3中不存在相互作用邊.
在圖1網絡中,蛋白質A,C,E分別為關鍵蛋白質,B,D,F分別為非關鍵蛋白質.經過動態網絡切分后,由于關鍵蛋白質往往具備保守性的原因,使得關鍵蛋白質更難在動態網絡中被改變,而非關鍵蛋白質的表達性更易受到外界的影響而發生變化,如蛋白質F在周期1,2,3中并未表達出“活性”.
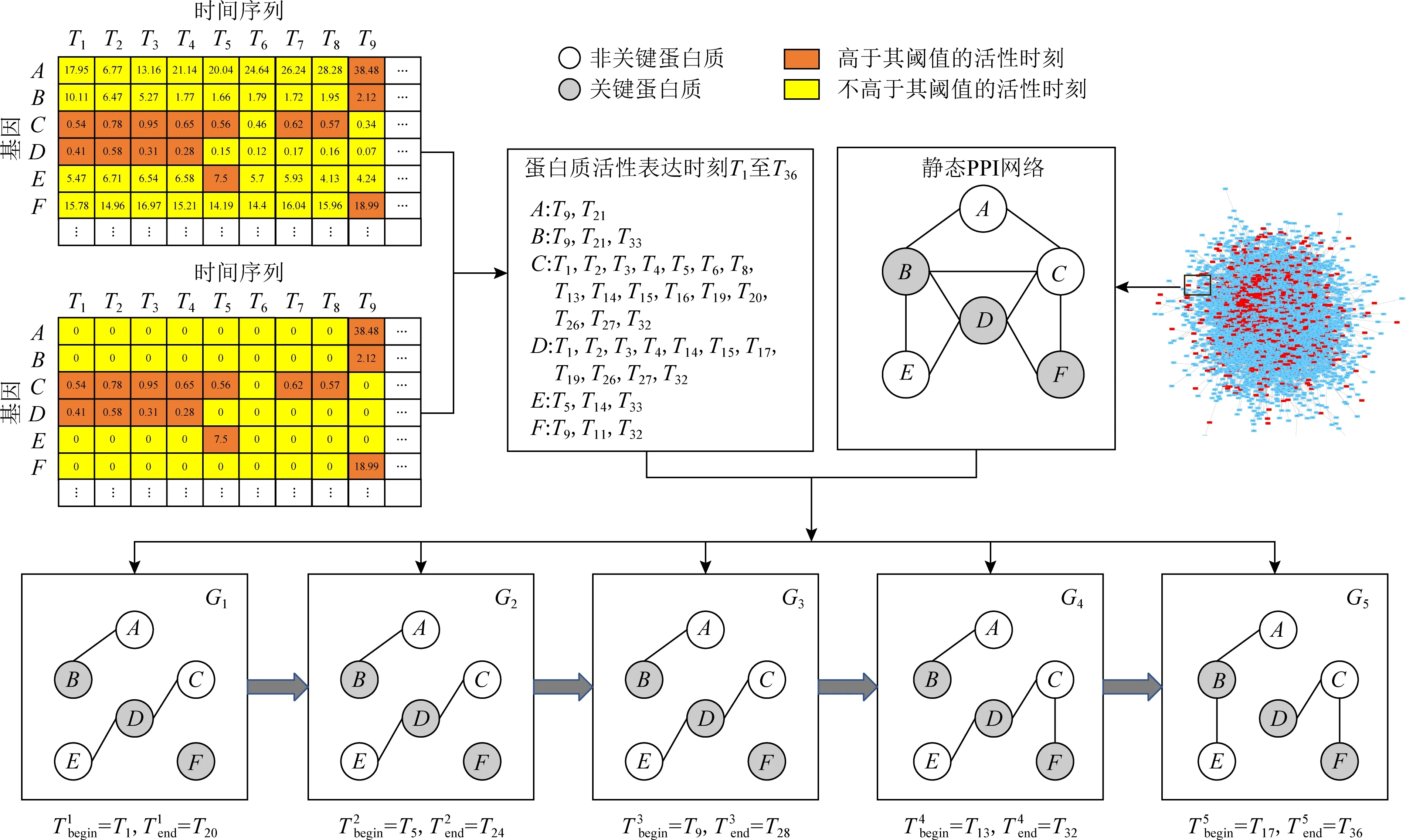
Fig. 1 Schematic diagram of dynamic network segmentation圖1 動態網絡切分示意圖
2.3 動態網絡切分中的關鍵蛋白質識別
由于動態網絡中蛋白質在不同周期下表達的“活性”不同,因此在計算最終關鍵蛋白質識別得分考慮子網中表達為“活性”的蛋白質出現次數.對比分析靜態PPI網絡和動態網絡切分下的關鍵蛋白質識別方法,其動態網絡切分下的蛋白質關鍵得分為:
(8)
其中,n(v)表示蛋白質v在k個子網中出現的次數,Mk(v)表示在第k個周期下的蛋白質v的關鍵得分,MMPN(v)表示蛋白質v在融合p個周期后的最終關鍵得分.
近年來,隨著對蛋白質相互作用網絡的不斷深入研究,許多基于網絡拓撲特征的方法和融合PPI網絡與生物信息的方法被提出.本文選取了9種基于靜態PPI網絡的關鍵蛋白質預測方法,其中包括7種網絡拓撲中心性方法DC,IC,EC,SC,BC,NC,LAC和2種融合基因表達方法PeC和WDC.通過對基因“活性”表達矩陣劃分p個周期,其對應的動態網絡切分方法分別為DPN,IPN,EPN,SPN,BPN,NPN,LPN,PPN,WPN.具體動態網絡切分的關鍵蛋白質預測方法公式如表1所示:

Talbe 1 Prediction Essential Proteins Equation for Converting Static PPI Network Method to Dynamic Network Segmentation Method表1 靜態PPI網絡方法轉換為動態網絡切分方法的預測關鍵蛋白質公式
描述計算動態網絡切分的算法在算法1中給出.
算法1.動態網絡切分算法.
輸入:蛋白質相互作用網絡G=(V,E)、基因表達數據V×T;
輸出:得分排名前q個關鍵蛋白質.
① 計算基因動態表達閾值:
for eachv∈Vdo
根據式(1)計算V(v);
根據式(2)計算S(v);
end for
② 構建基因“活性”表達矩陣:
for eachv∈Vdo
for eacht∈Tdo
end for
end for
根據式(4)構建矩陣X′;
③ 構建動態網絡子網:
for eachk∈[1,p] do
for eache(v,u)∈Edo
根據式(7)計算ek(v,u);
end for
end for
end for
④ 計算各子網中蛋白質得分:
for eachk∈[1,p] do
for eachv∈Vdo
根據表1計算Mk(v);
end for
end for
⑤ 計算蛋白質最終得分:
for eachv∈Vdo
根據式(8)計算MMPN(v);
end for
⑥ 根據MMPN(v)得分降序排列,取前q個蛋白質作為關鍵蛋白質輸出.
動態網絡切分算法主要由6部分組成:第1步計算基因動態閾值,時間復雜度為O(n);第2步循環各蛋白質各時間點的表達量,構建基因“活性”表達矩陣,時間復雜度為O(nT);第3步對“活性”表達矩陣進行切分,并結合靜態PPI網絡構建動態網絡子網,時間復雜度為O(meT′);第4步計算各子網中蛋白質得分,時間復雜度為O(mn);第5步對各個子網中的得分進行累加求和再除以蛋白質節點在動態網絡中出現的次數,以此作為蛋白質最終得分,時間復雜度為O(n);第6步根據最終得分降序排列,輸出前q個蛋白質作為預測的關鍵蛋白質.總時間復雜度為O(n+nT+meT′+mn+n).其中,n代表蛋白質節點的個數,m代表切分子網的個數,T代表基因周期長度,T′代表切分后的基因周期長度.
3 實驗數據
由于酵母蛋白質網絡和關鍵蛋白質數據是相對比較完善的,因此本文采用釀酒酵母(saccharomyces cerevisiae)來進行實驗.另外,本文還采用了大腸桿菌(Escherichia coli, E.coli)和人類膀胱部位(bladder)的數據來進一步驗證實驗.
酵母和大腸桿菌的PPI網絡數據下載自DIP數據庫,丟棄掉網絡中的重復相互作用和自我相互作用,最終釀酒酵母的PPI網絡包含了5 093個蛋白質和24 743個相互作用,大腸桿菌的PPI網絡包含了2 727個蛋白質和11 803個相互作用.人類膀胱的PPI網絡數據從BioGRID(Version 3.5.182)下載得到,包含1 748 436條相互作用,去除重復和自環之后包含15 721個基因和322 406個相互作用.
基因表達數據從基因表達綜合數據庫(GEO)中獲取.酵母的基因表達數據下載自GESE3431,包含6 777個基因產物和36個時間點,其中有4 858個基因參與釀酒酵母PPI網絡.大腸桿菌表達數據在GSE3905中,包含7 312個基因產物和8個時間點.人類膀胱的表達數據在GSE86354中,提供了基因型-組織表達(GTEx)項目產生的8個組織位點的1 558份樣本的表達譜,其中膀胱包含了11個時間點.
關鍵蛋白質數據通過整合MIPS[30],SGD[31],DEG[32],SGDP[33]四個數據庫,其中釀酒酵母的關鍵蛋白質有1 285個,其中有1 167個蛋白質出現在釀酒酵母PPI網絡中.大腸桿菌在其PPI網絡中包含254個關鍵蛋白質.人類膀胱的關鍵基因數據在在線關鍵基因數據庫(OGEE)(downloaded at 20/10/2020)中下載得到21 556個基因座,在Uniprot網站上將其進行映射對應的18 900個基因,包含7 123個關鍵基因.實驗數據集及代碼提交至開源網站:https://github.com/jczhongcs/DevideDynamicNetwork.
4 實驗結果與分析
4.1 Top排序分析
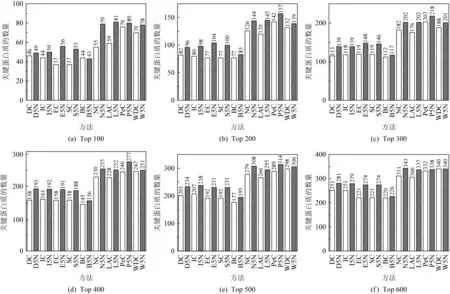
Fig. 2 Top ranking number analysis of essential proteins by dynamic network segmentation and other prediction methods in yeast圖2 酵母中動態網絡切分方法與其他方法在關鍵蛋白質預測的Top排序數量分析
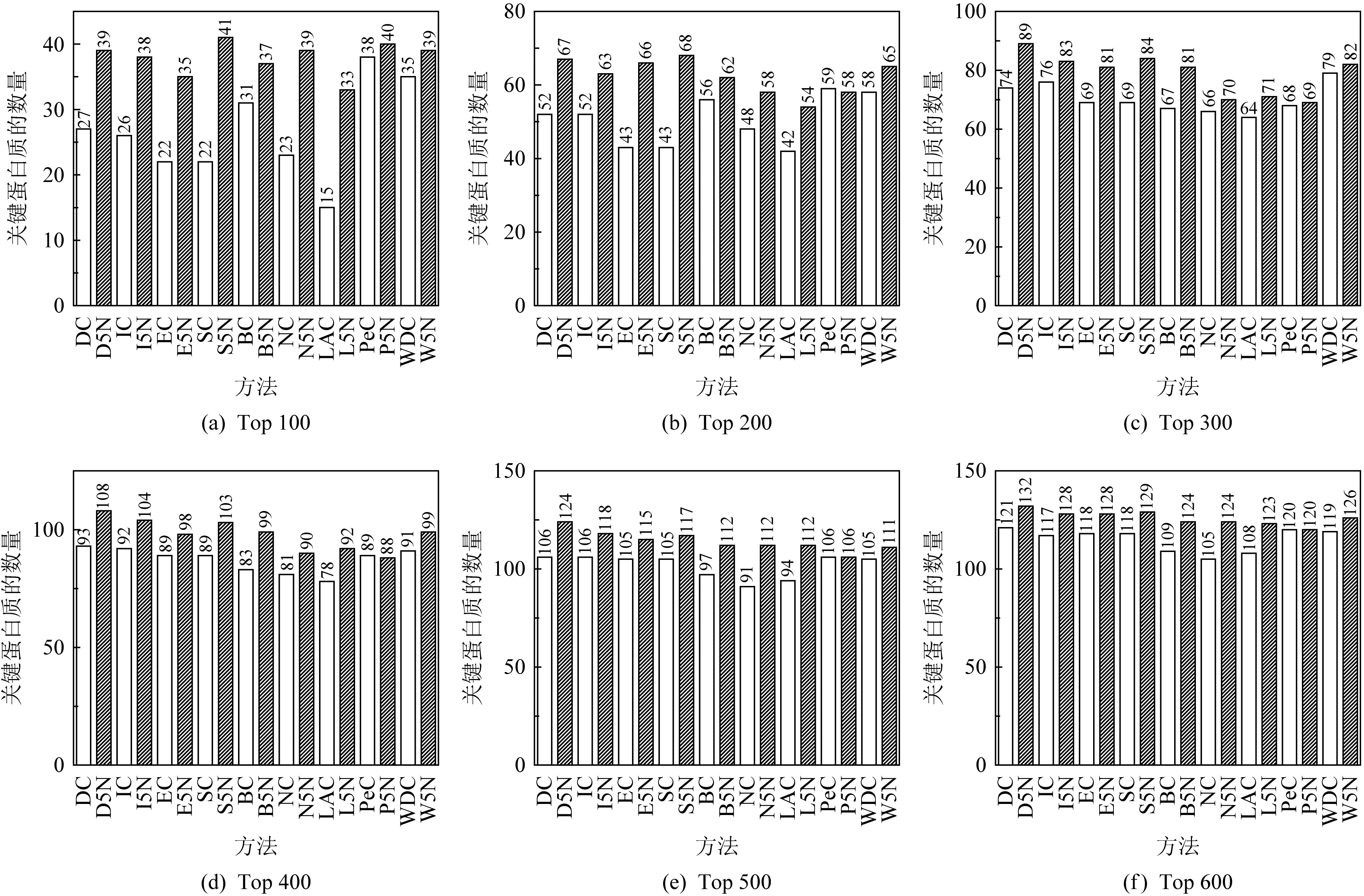
Fig. 3 Top ranking number analysis of essential proteins by dynamic network segmentation and other prediction methods in E. coli圖3 大腸桿菌中動態網絡切分方法與其他方法在關鍵蛋白質預測的Top排序數量分析
本文對比多個參數實驗結果,將設置u=10,p=5.對基因表達周期切分為5個周期,當前周期與下一個周期的間隔占總周期長度的1/10.為了充分體現經過5個周期的動態網絡切分方法的優越性,本文將7種中心性方法(DC,IC,EC,SC,BC,NC,LAC)以及2種融合性方法(PeC和WDC),與其在動態網絡切分后的預測方法(D5N,I5N,E5N,S5N,B5N,N5N,L5N,P5N,W5N)的預測結果進行比較.選擇其預測的排名Top 100,Top 200,Top 300,Top 400,Top 500,Top 600的關鍵蛋白質,并判斷其中所包含的正確的關鍵蛋白質數量.酵母與大腸桿菌的Top分析結果如圖2、圖3所示.在酵母預測得分的Top 100個蛋白質中,中心性方法DC,IC,EC,SC,BC,NC,LAC,PeC,WDC分別預測了46,44,37,37,44,55,59,76,70個正確的關鍵蛋白質;D5N,I5N,E5N,S5N,B5N,N5N,L5N,P5N,W5N分別預測了49,50,56,53,43,79,81,80,78個正確的關鍵蛋白質.除B5N外,經過動態網絡切分之后的方法在得分Top 100個蛋白質中所預測的正確關鍵蛋白質數量都要大于原靜態網絡中的方法,其中L5N預測的關鍵蛋白質達81個,識別率為所有方法中最高.雖然B5N在Top 100中比BC少預測了一個關鍵蛋白質,但在Top 200,Top 300,Top 400,Top 500,Top 600中預測的關鍵蛋白質都要高于原方法BC.在大腸桿菌中關鍵蛋白質的Top分析中可以看出,在Top 100中D5N,I5N,E5N,S5N,B5N,N5N,L5N,P5N,W5N分別預測了39,38,35,41,37,39,33,40,39個,相比于原方法分別高出了12,12,13,19,6,16,18,2,4個,其中S5N預測了所有方法中最多的關鍵蛋白質數量.在之后的Top 200,Top 300,Top 400,Top 500,Top 600的Top分析中,經過動態網絡切分后的中心性方法相比融合性方法識別的關鍵蛋白質也更多.從識別結果中可以看出,中心性方法與融合方法在動態網絡切分后都能預測出更多的關鍵蛋白質,這表明經過動態網絡切分的方法相比靜態PPI網絡可以有效地提取各個周期中關鍵蛋白質的活性信息,而關鍵蛋白質更多地參與細胞中重要的生命活動,相比非關鍵蛋白質具有更高的活性.因此,動態網絡切分的方法對關鍵蛋白質的識別擁有更高的準確性.同時,一方面動態網絡切分對基因活性表達矩陣中不高于閾值的表達量置0,降低了處于非活性狀態中的基因表達值的影響,提高了活性表達量的可靠性,有助于進一步過濾網絡中的假陰性及假陽性的噪聲數據.另一方面也側面說明了引入基因隨周期表達的概念可以有效地挖掘各個周期中不同活性表達水平的關鍵蛋白質.
4.2 基于ROC曲線分析和多種性能評估
受試者工作特征ROC曲線常用來評估二分類系統的好壞,縱坐標表示真陽性率,橫坐標表示假陽性率,曲線上每一個點反映對同一信號刺激性的感受性.本文選取動態網絡切分下的代表性方法W5N與其他方法進行比較.酵母與大腸桿菌的ROC曲線如圖4和圖5所示:
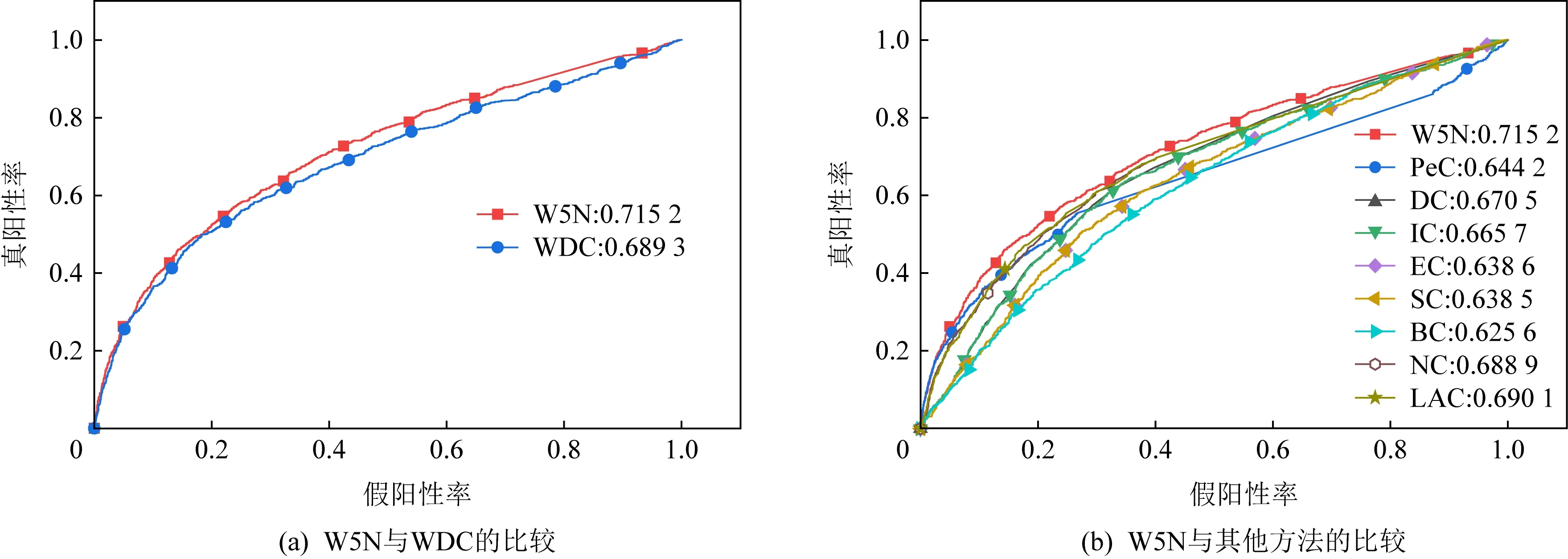
Fig. 4 ROC curve and AUC value of dynamic network segmentation method and other prediction methods in yeast圖4 酵母中動態網絡切分方法與其他預測方法的ROC曲線和AUC值
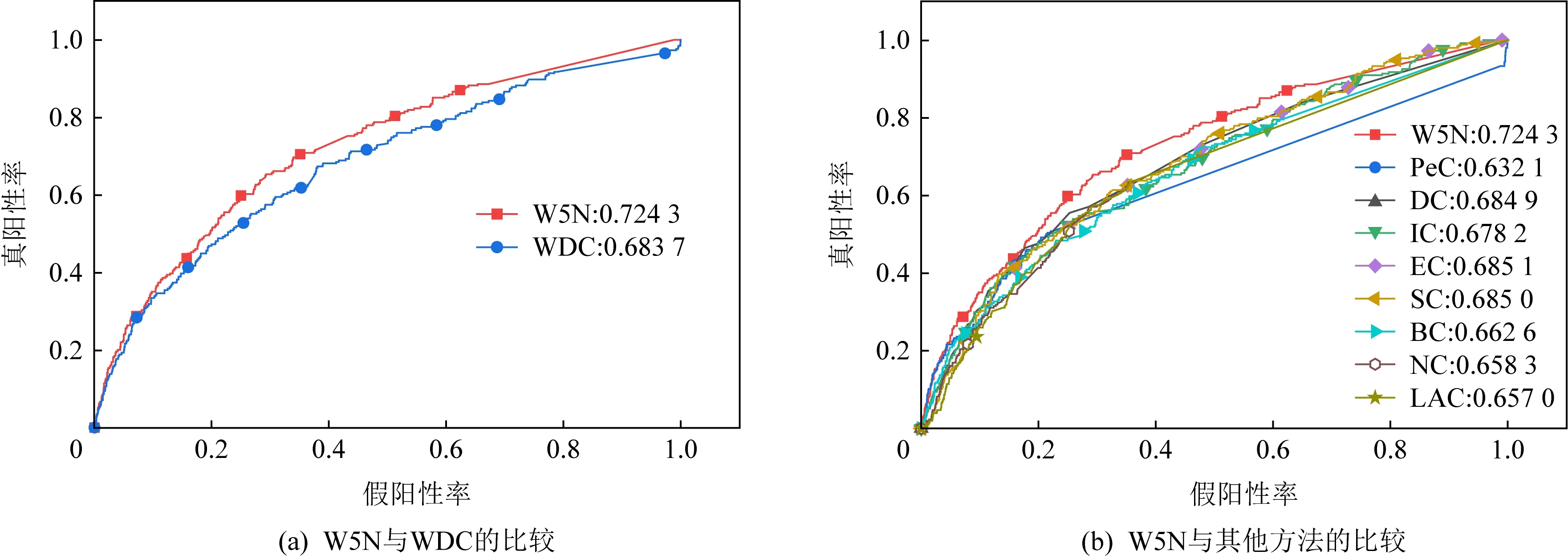
Fig. 5 ROC curve and AUC value of dynamic network segmentation method and other prediction methods in E.coli.圖5 大腸桿菌中動態網絡切分方法與其他預測方法的ROC曲線和AUC值
如圖4酵母ROC曲線所示,在其經過5個周期切分之后的動態網絡中的W5N與原方法WDC的曲線下面積AUC分別為0.715 2和0.689 3,W5N高出了0.025 9;其他方法PeC,DC,IC,EC,SC,BC,NC,LAC的AUC分別為0.715 2,0.670 5,0.665 7,0.638 6,0.638 5,0.625 6,0.688 9,0.690 1,與之相比,W5N為所有方法中ROC曲線下面積最大.從圖4說明,動態網絡切分后的方法相比原方法能區分出更多的關鍵蛋白質與非關鍵蛋白質,使網絡變得更為可靠.
如圖5所示,在大腸桿菌ROC曲線中,W5N曲線下面積AUC為0.724 3,WDC,PeC,DC,IC,EC,SC,BC,NC,LAC分別為0.683 7,0.632 1,0.684 9,0.678 2,0.685 1,0.685 0,0.662 6,0.658 3,0.657 0,W5N的AUC為所有方法最高.這表明經過動態網絡切分后的W5N能預測出更多的關鍵蛋白質.
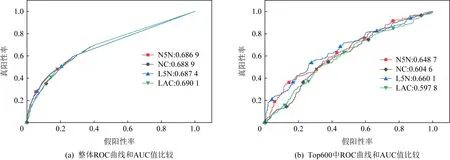
Fig. 6 ROC curve and AUC value of N5N and L5N, NC and LAC in yeast圖6 酵母中N5N,L5N與NC,LAC的ROC曲線和AUC值
為了進一步驗證動態網絡切分對預測關鍵蛋白質性能的提升,本文選取酵母預測得分前1 167個蛋白質和大腸桿菌預測得分前254個蛋白質作為預測的關鍵蛋白質,并利用ROC曲線下面積AUC、敏感性(SN)、特異性(SP)、假陽性率(FPR)、陽性預測值(PPV)、陰性預測值(NPV)、F-measure、準確度(ACC)和Matthews相關系數(MCC)這9個指標來對各個方法的性能進行評估.
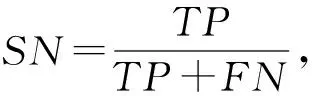
(9)
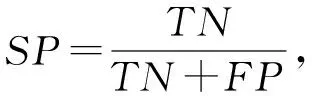
(10)
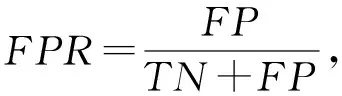
(11)
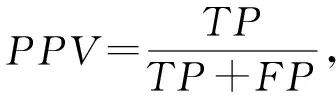
(12)
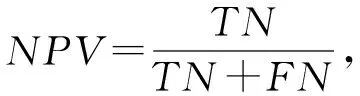
(13)
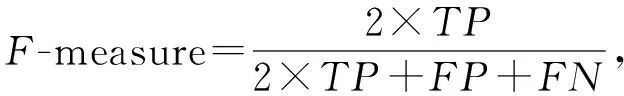
(14)
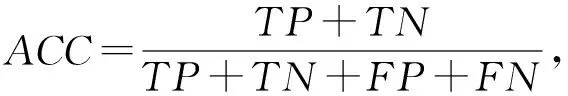
(15)
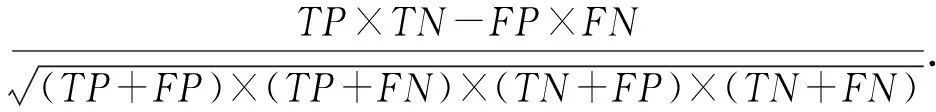
(16)
式(9)~(16)中,真陽性TP表示預測正確的關鍵蛋白質;真陰性TN表示預測正確的非關鍵蛋白質;假陽性FP表示關鍵蛋白質錯誤地被預測為非關鍵蛋白質;假陰性FN表示非關鍵蛋白質錯誤地被預測為關鍵蛋白質.
由于N5N和L5N整體的AUC略低于NC和LAC,本文又分別計算了N5N,NC,L5N,LAC預測得分Top 600的蛋白質的AUC,結果如圖6所示.從圖6的ROC曲線圖不難看出,在研究人員更關心的預測排名靠前的ROC下曲線面積中,N5N,NC,L5N,LAC的AUC值分別為0.648 7,0.604 6,0.660 1,0.597 8,這也說明N5N和L5N在預測得分排名越高的情況下,相比NC和LAC的預測得分結果更為可靠.由表2可知,經過動態網絡切分后的大部分方法的AUC相比原方法都得到了提升,其中W5N的AUC達到了最高的0.715 2.在其余的統計指標中,除B5N的指標略低于原有方法BC指標之外,W5N,P5N,D5N,S5N,E5N,I5N,N5N,L5N的各項指標全都超過了原有方法.這說明在酵母中進行動態網絡切分的方法可以提升在靜態PPI網絡中方法的性能.其中,酵母中W5N的SN,SP,PPV,NPV,F-measure,ACC,MCC分別為0.474 7,0.843 9,0.474 7,0.843 9,0.474 7,0.759 3,0.318 6,假陽性率FPR為最低的0.156 1,各項指標均在同類型方法WDC和其他所有方法中達到了最高,這說明了W5N在預測酵母的關鍵蛋白質中的識別率最高、性能最好.
由表3可知,在大腸桿菌中的動態網絡切分中的各項方法AUC相比于原有方法均有較大提升,其中S5N的AUC達到了最高的0.729 7.在其他指標中,其中D5N的SN,SP,PPV,NPV,F-measure,ACC,MCC分別為0.315 0,0.929 6,0.315 0,0.929 6,0.315 0,0.872 4,0.244 6,均為所有方法中最高,FPR為最低的0.070 4.各項方法經過動態網絡切分后,相比原方法的指標都得到了提升,使預測結果更為可靠.
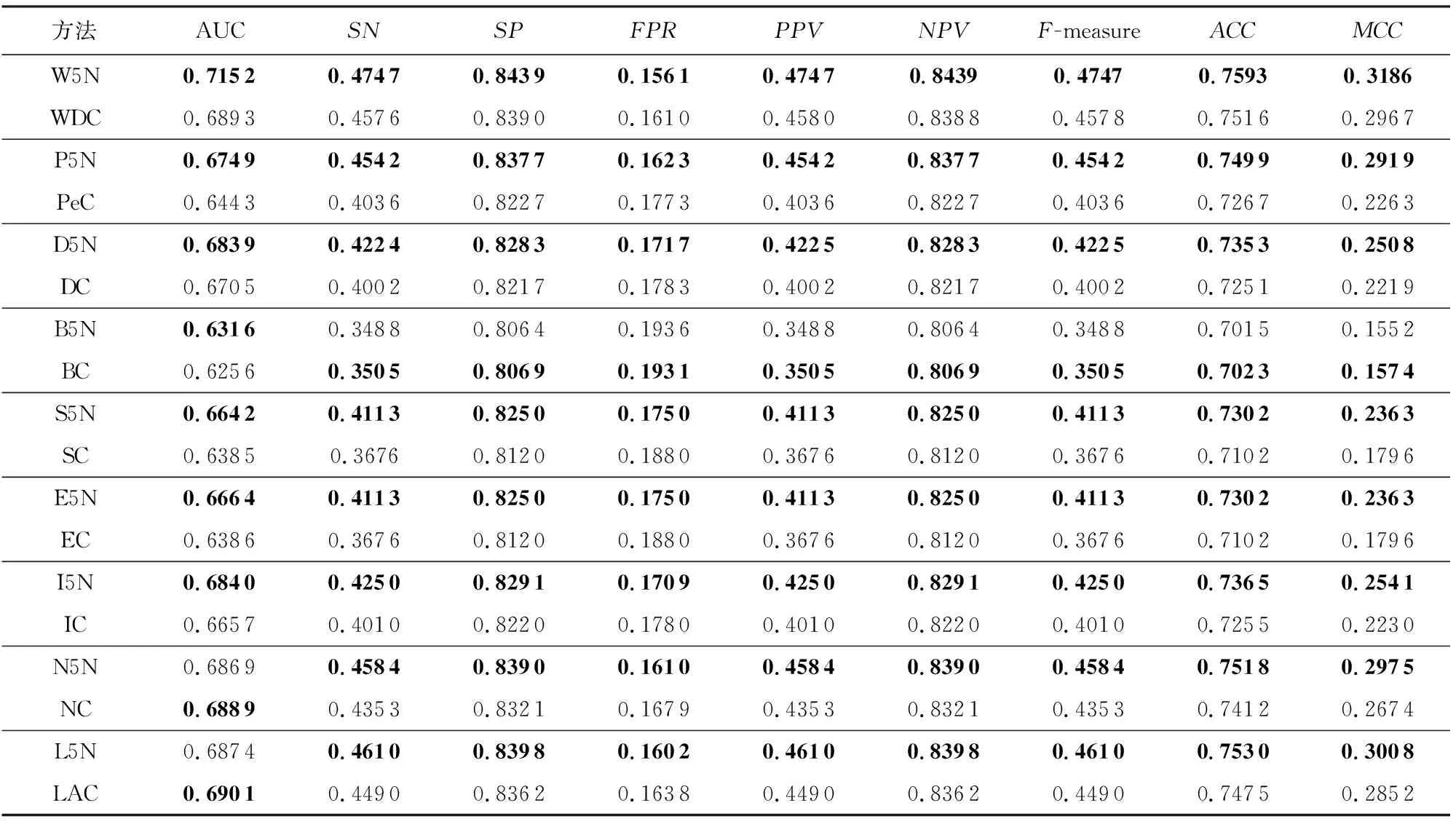
Table 2 Evaluation and Analysis of Yeast表2 酵母評估分析
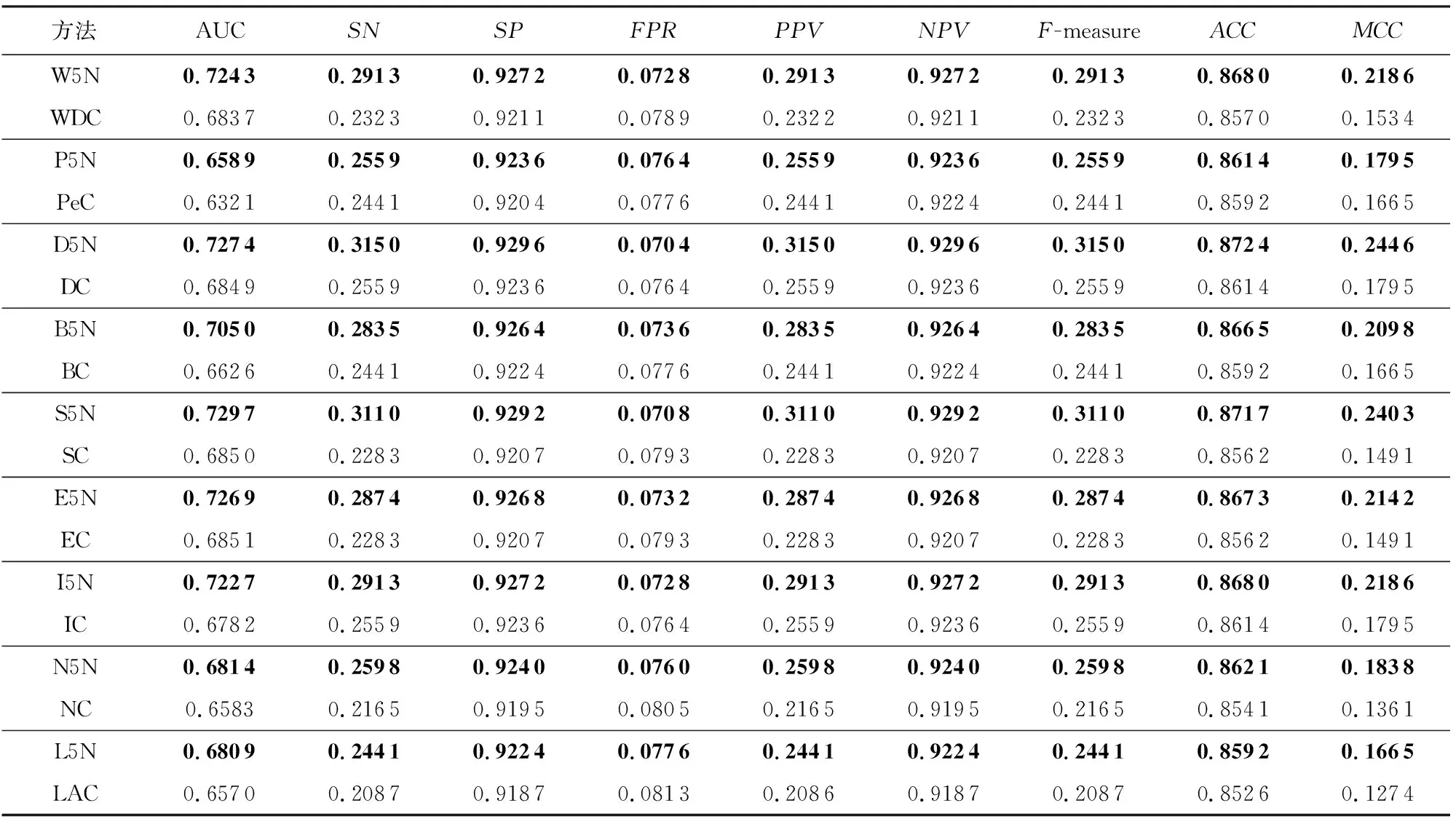
Table 3 Evaluation and Analysis of E.coli表3 大腸桿菌評估分析
4.3 基于重疊性和Jackknife分析
為了進一步分析動態網絡切分方法在酵母與大腸桿菌中預測關鍵蛋白質的表現,本文將選取動態網絡切分中的代表性方法W5N與其余9種關鍵蛋白質預測方法(DC,EC,SC,BC,IC,NC,LAC,PeC,WDC)預測得分的Top 100個關鍵蛋白質進行重疊分析.酵母中W5N與其他預測方法之間重疊數量如表4所示,大腸桿菌中W5N與其他預測方法之間重疊數量如表5所示.以表4中的W5N為例:Ci為其他關鍵蛋白質預測方法,CW5N∩Ci表示W5N和其余各關鍵蛋白質預測方法之間識別關鍵蛋白質的重疊部分數量,|Ci-CW5N|表示由W5N和其余各關鍵蛋白質預測方法識別關鍵蛋白質的非重疊部分數量.

Table 4 The Number of Overlaps Between W5N and Other Methods in Yeast表4 酵母中W5N與其他方法之間的重疊數量
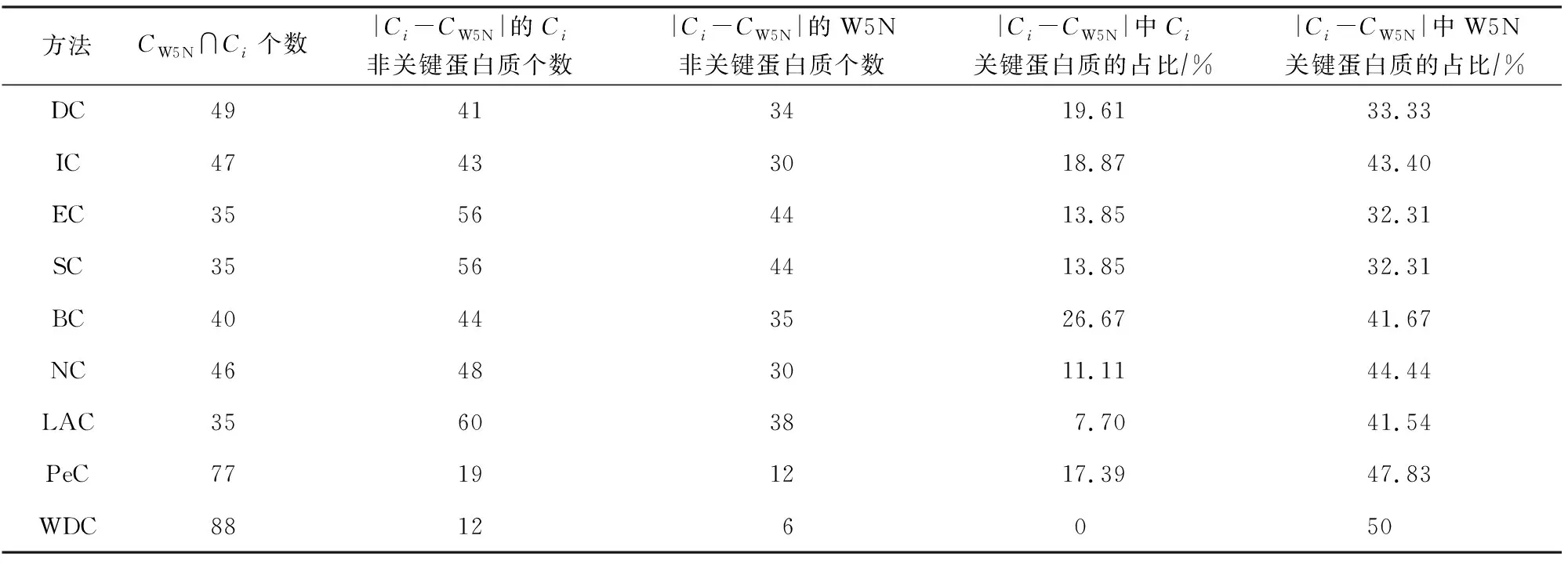
Table 5 The Number of Overlaps Between W5N and Other Methods in E.coli表5 大腸桿菌中W5N與其他方法之間的重疊數量
從表4可以看出,在酵母中W5N與中心性方法(DC,EC,SC,BC,IC,NC,LAC)預測了較少的相同關鍵蛋白質,其中W5N與DC和NC的重疊部分最高,預測了41個相同的關鍵蛋白質,在其非重疊部分,W5N預測的關鍵蛋白質百分比超過了80%.W5N與融合了生物信息的PeC和WDC相比有較高的重疊,但在非重疊部分預測的關鍵蛋白質更多,其中與PeC的非重疊部分的關鍵蛋白質達到了82%,這說明使用動態網絡切分方法預測關鍵蛋白質是很有必要的.在W5N與其他方法的重疊部分中預測的非關鍵蛋白質更少,預測的關鍵蛋白質更多.以SC為例,SC的|Ci-CW5N|的數量為77,在該77個非重疊蛋白質中,SC識別的關鍵蛋白質占比為24.68%,而W5N識別的關鍵蛋白質占比為75.32%,說明在非重疊部分,W5N相比SC多識別了超過50%的關鍵蛋白質,同時也反映了W5N識別關鍵蛋白質的可靠性.
從表5可以看出,由于大腸桿菌相比于酵母的關鍵蛋白質占總體蛋白質的比例較低,關鍵蛋白質的數量較少,非關鍵蛋白質數量較多,所以造成預測的關鍵蛋白質占比相比酵母較低.經過動態網絡切分后的W5N與中心性方法的重疊部分較低,與PeC和WDC的重疊部分較高.其中與原方法WDC的重疊部分最高為88個,但在非重疊部分的12個蛋白質中,WDC錯誤地將非關鍵蛋白質全部預測為關鍵蛋白質,而W5N預測正確的個數為6個.且W5N與其他所有方法的非重疊部分相比,預測正確的關鍵蛋白質全部高于其余對比方法.這也表明了W5N在大腸桿菌中能更好地識別關鍵蛋白質.
為了更加細致地分析動態網絡切分方法的優越性,本文引入Jackknife方法對其分析.橫軸表示預測為關鍵蛋白質的數量,縱軸表示在預測為關鍵蛋白質的數量中真實的蛋白質數量.曲線下面積越大,表明預測的關鍵蛋白質數量越多.酵母和大腸桿菌中W5N與P5N的Jackknife曲線分別如圖7和圖8所示:
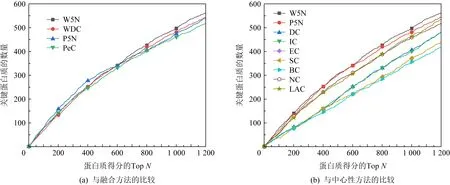
Fig. 7 Jackknife overlap curve analysis of each method in yeast data圖7 酵母數據中各方法的Jackknife重疊曲線分析
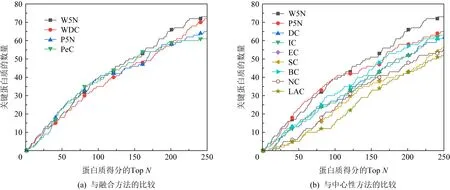
Fig. 8 Jackknife overlap curve analysis of each method in E.coli data圖8 大腸桿菌數據中各方法的Jackknife重疊曲線分析
從圖7和圖8可以看出,圖7(a)和圖8(a)中P5N識別的關鍵蛋白質數量在前段都處在最高位置,隨著預測排名的增加,W5N開始處于最高位置,識別的關鍵蛋白質數目超過了P5N和其他方法.這說明P5N,W5N的關鍵蛋白質識別率都在分別優于原方法PeC和WDC的同時,全部高過靜態網絡中心性的方法,從而說明經過動態網絡切分后的方法對識別關鍵蛋白質的有效性和優異性.
4.4 參數分析
本文進一步分析不同的基因活性表達矩陣閾值和不同的基因活性表達矩陣的周期劃分對于預測性能的影響.首先,針對酵母和大腸桿菌數據,采用不同的基因活性表達矩陣閾值參數,分別設置為1,1.5,2,2.5,3來分析對識別關鍵蛋白質的影響程度,結果如附錄A的表A1、表A2所示.由酵母實驗結果可見,在設置的5種閾值參數中,用于識別關鍵蛋白質的9種方法在不同參數設置的結果略有差別.在參數為2時,所構建的基因活性表達矩陣中AUC值均相對最高;當參數為2.5時,在Top 100至Top 600分析時呈現出較好的結果.大腸桿菌的基因表達數據經過5個閾值系數過濾得到5個基因活性表達矩陣,這5個基因活性表達矩陣的差別極小,劃分后的子網的邊對應關系均完全一致,即中心性結果一致,最終分別計算出的5個P5N與W5N相差極小,最終5個閾值系數在9種方法中所得出的Top結果一致.
此外,本文設定不同的基因活性表達矩陣的周期劃分,將基因活性表達矩陣分別劃分為3,4,5,6,7個周期來測試周期的劃分對實驗的影響,結果如附錄A表A3、表A4所示.由酵母與大腸桿菌的實驗結果表明,基因活性表達矩陣的周期劃分對基于中心性方法關鍵蛋白質的識別影響較小,在AUC值分析和Top結果上略有差別.
4.5 基于動態網絡分析對比
在之前的實驗中,本文對比了動態網絡切分識別方法與靜態網絡中識別方法.實驗結果表明,經過動態網絡的切分關鍵蛋白質的識別率可以得到有效的提升.為了進一步討論動態網絡切分對預測關鍵蛋白質的有效性,本文還將動態網絡切分方法與動態網絡NF-PIN的識別結果進行對比分析.
NF-PIN動態網絡在進行酵母實驗時,將基因表達數據中的36個時間點分為12個時間段.本文為了單獨比較方法的預測性能,將選取與NF-PIN一致的基因表達數據與PPI網絡進行方法評估,實驗結果如表6所示.由于大腸桿菌的基因表達數據中包含的時間點為8個,在動態網絡中難以形成5個有效的周期,鑒于此類情況,本文實驗獲取了與酵母NF-PIN動態網絡中時間相仿的人類膀胱的數據集進行實驗,人類膀胱的時間點為11個,實驗結果如表7所示.
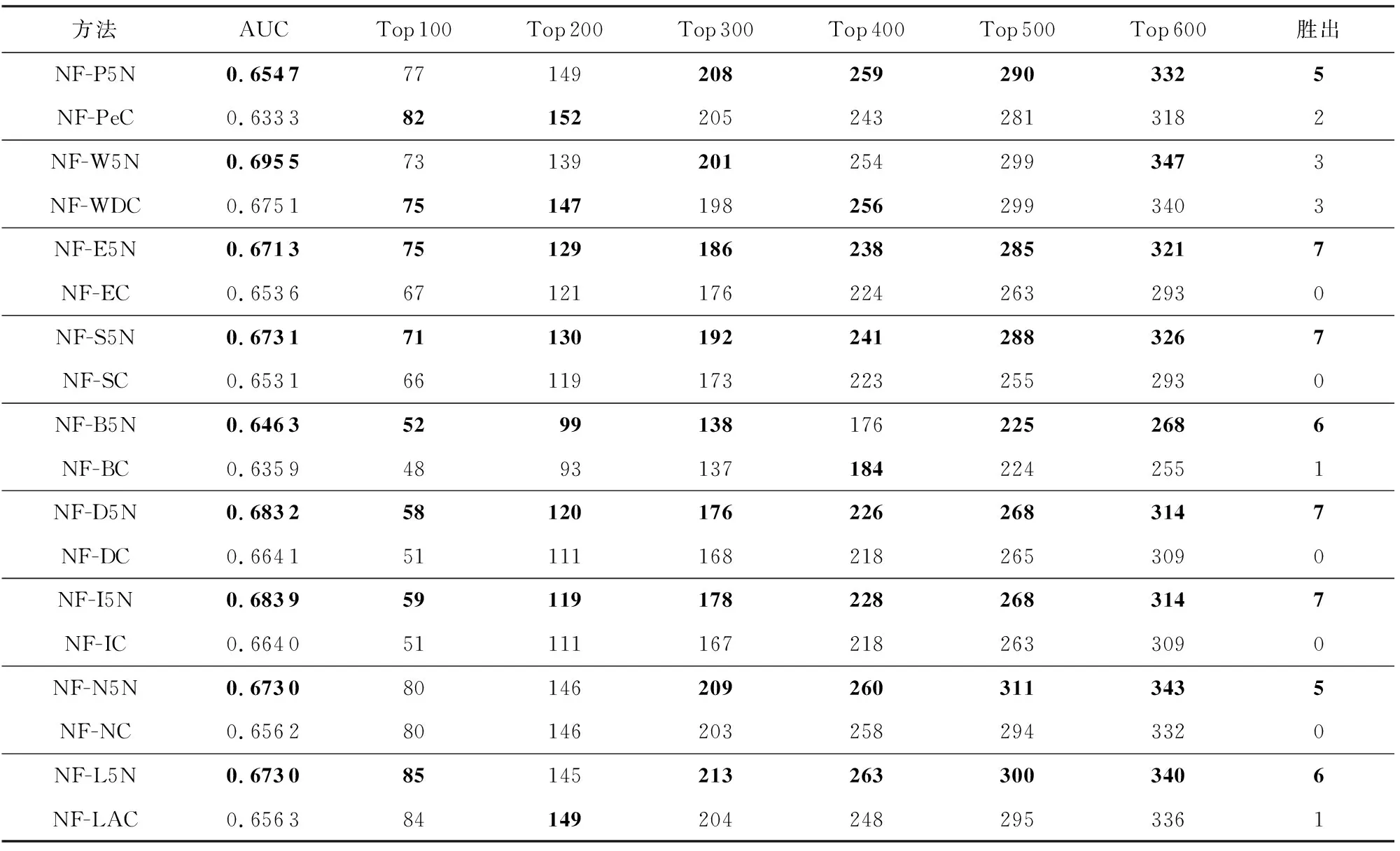
Table 6 Analysis of Various Prediction Methods for Dynamic Network Segmentation andNF-PIN Dynamic Network in Yeast Data表6 酵母數據中動態網絡切分方法與NF-PIN動態網絡各種預測方法分析
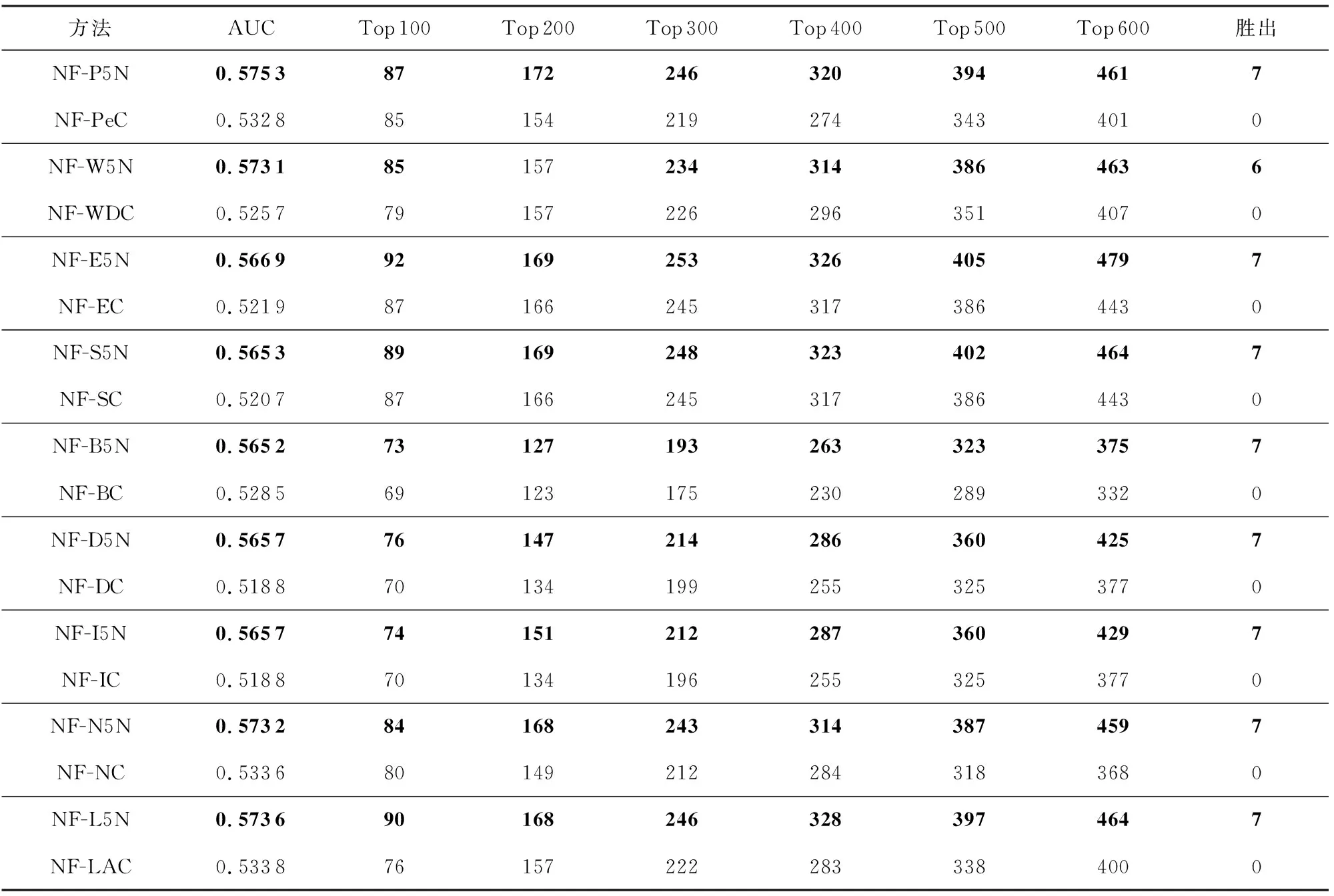
Table 7 Analysis of Various Prediction Methods for Dynamic Network Segmentation andNF-PIN Dynamic Network in Human Bladder Data表7 人類膀胱數據中動態網絡切分方法與NF-PIN動態網絡各種預測方法分析
從表6中可以看出,在動態網絡切分與動態網絡NF-PIN輸入了一致數據的實驗中,所有經過切分之后的方法相比原方法,其AUC均得到了一定程度的提升.其中NF-W5N的AUC達到了最高的0.695 5,相比NF-PIN中AUC最高的0.675 1還要高出2.04個百分點,這表明由動態網絡切分方法預測的關鍵蛋白質準確度更高.在中心性方法Top分析中,動態網絡切分方法預測出的關鍵蛋白質數量都基本超過了未經切分的動態網絡NF-PIN,其中NF-E5N,NF-S5N,NF-D5N,NF-I5N,NF-N5N相比NF-EC,NF-SC,NF-DC,NF-IC,NF-NC的識別率有全面提升.其中,NF-L5N在研究人員最關注的Top 100中預測了最多的85個關鍵蛋白質.這表明動態網絡NF-PIN利用生物表達的動態性,在識別關鍵基因中有著較高的準確度,而動態網絡切分在利用動態性的基礎上進一步結合了生物周期性表達的特性,降低了網絡中來自假陽性及假陰性數據的影響,相比僅利用基因表達動態性的動態網絡能夠有效識別出更多的關鍵蛋白質.
在表7中可以看出,在人類膀胱的數據集中,所有動態網絡切分方法的AUC都要高于原方法.其中,NF-P5N在預測關鍵基因中AUC達到了最高的0.575 3.從Top分析中可以看出,所有動態網絡切分方法相比原方法都得到了較大的提升,其中NF-E5N在Top 100中預測了最多的92個關鍵基因.實驗結果表明,在人類膀胱數據集中,經過切分后的網絡在各個方法上都比動態網絡NF-PIN的識別率要高.這也說明了融合5個周期的動態網絡相比于不切分的動態網絡擁有更好的性能,能識別出更多的正確結果.
5 總 結
本文融合基因表達數據中的時序數據擴展了PPI網絡的動態性.實驗通過對酵母、大腸桿菌及人類膀胱的蛋白質數據的分析探索,將靜態PPI網絡劃分了多個周期及構建融合多個子網信息的動態網絡,以盡可能避免靜態網絡中假陽性和假陰性數據的影響,最大限度地提取在蛋白質隨環境變化時所具有的保守性.實驗結果表明:在酵母、大腸桿菌及人類膀胱中進行動態網絡切分的方法可以有效地提高關鍵蛋白質的識別率,識別出更多的關鍵蛋白質.
作者貢獻聲明:鐘堅成和潘毅提出研究思路和實驗方案,以及對論文的修改進行審查;鐘堅成和彭瑋進行研究目標分析和研究方案總結;方卓負責實驗推進和論文初稿撰寫;瞿佐航負責數據整理和論文校對;鐘穎參與實驗測試.
猜你喜歡
中學生數理化(高中版.高考理化)(2021年6期)2021-07-28 06:21:04
人大建設(2019年9期)2019-12-27 09:06:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
NBA特刊(2014年7期)2014-04-29 00:44:03
中國商人(2013年1期)2013-12-04 08:52:52
兒童時代(2009年5期)2009-05-21 05:31:26
