基于信息熵權法優(yōu)選香果健消片揮發(fā)油羥丙基-β-環(huán)糊精包合工藝及其表征
2022-07-07 09:22:44郝佳旭查麗春崔利利高家菊曾鵬輝武旭潤馬云淑
中草藥 2022年13期
郝佳旭,查麗春,范 曉,崔利利,高家菊,曾鵬輝,武旭潤,馬云淑*
基于信息熵權法優(yōu)選香果健消片揮發(fā)油羥丙基-β-環(huán)糊精包合工藝及其表征
郝佳旭1, 2, 3,查麗春1, 2, 3,范 曉1, 2, 3,崔利利1, 2, 3,高家菊1, 2, 3,曾鵬輝1, 2, 3,武旭潤1,馬云淑1, 2, 3*
1. 云南中醫(yī)藥大學中藥學院,云南 昆明 650500 2. 云南省高校外用給藥系統(tǒng)與制劑技術研究重點實驗室,云南 昆明 650500 3. 云南省傣醫(yī)藥與彝醫(yī)藥重點實驗室,云南 昆明 650500
優(yōu)選羥丙基-β-環(huán)糊精(2-hydroxypropyl-β-cyclodextrin,HPCD)包合香果健消片(Xiangguo Jianxiao Pian,XJP)揮發(fā)油最佳工藝。在單因素實驗基礎上,以包合率、收率、含油率、每克包合物中1,8-桉葉素的含量為指標,HPCD與無水乙醇的質量體積比、包合溫度、包合時間為影響因素,采用信息熵權法確定各指標權重系數(shù),計算綜合評分,建立正交試驗優(yōu)選最佳包合工藝。采用薄層色譜、紫外吸收光譜、傅里葉紅外光譜、差示掃描量熱法、掃描電鏡、X射線衍射、GC-MS等方法對包合物進行表征,并考察其穩(wěn)定性影響因素。最佳包合工藝為HPCD與無水乙醇的質量體積比為12∶20(g/mL),包合溫度55 ℃,包合時間2 h,經驗證制得的包合物平均包合率為88.15%、收率為96.11%、含油率為7.65%、包合物中1,8-桉葉素的含量為20.28 mg/g。揮發(fā)油成功進入HPCD空腔中,包合前后揮發(fā)油成分基本未發(fā)生變化,包合物穩(wěn)定性較好。優(yōu)選的揮發(fā)油包合工藝穩(wěn)定可行。
香果健消片;揮發(fā)油;羥丙基-β-環(huán)糊精;1,8-桉葉素;信息熵權法;正交試驗;包合工藝
香果健消片(Xiangguo Jianxiao Pian,XJP)為中藥部頒標準品種,原方為蜘蛛香(炒焦)250 g、草果(去殼、炒焦)100 g、木香(炒)70 g、糯米80 g。方中蜘蛛香辛溫散寒,具有消食健胃、理氣止痛的作用,善治胃腸疾病,為君藥。
蜘蛛香在我國少數(shù)民族中有著廣泛的應用,如白族、布依族等使用蜘蛛香水煎液治療腹痛、腹脹、疳積等;阿昌族、傣族將蜘蛛香用作食療,主治小兒消化不良[1]。現(xiàn)代藥理研究表明,蜘蛛香通過下調磷脂肌醇-3-激酶(phosphatidylinositol-3-kinase,PI3K)以及蛋白激酶B(proteinkinase B,Akt)的表達水平,并通過抑制脫水來治療由輪狀病毒引起的腹瀉[2]。而在歐洲,蜘蛛香更常用于治療抑郁癥,有研究指出,蜘蛛香中總環(huán)烯醚萜類化合物可以通過調節(jié)腦-腸軸,改善大鼠慢性不可預知性刺激所致抑郁癥[3]。
木香與草果行氣止痛、燥濕溫中、消食導滯,共為臣藥,以加強蜘蛛香理氣止痛之功效,又能導滯健脾。木香中的木香烴內酯和去氫木香內酯以及乙醇提取物均對胃潰瘍有著顯著療效[4-5]。而草果水提物則可以改善洛哌丁胺誘導的小鼠便秘癥狀。可見XJP以調暢氣機、疏通經脈為治則,善治因氣機壅遏、氣滯不通所致小兒腹痛。
方中蜘蛛香、草果、木香3味藥材均具有揮發(fā)油類成分,且揮發(fā)油成分是其重要的活性成分。蜘蛛香揮發(fā)油以萜類成分為主,具有抗菌、抗氧化作用[7]。木香揮發(fā)油中主要成分為去氫木香內酯[8],同樣具有抗菌、抗氧化作用[9-10]。草果揮發(fā)油主要含有1,8-桉葉素、α-蒎烯等成分,具有抗氧化、調節(jié)腸胃功能、抗菌、抗腫瘤等作用[11]。在制劑過程中,揮發(fā)油傳統(tǒng)加入方式為直接加入,由于揮發(fā)油理化性質不穩(wěn)定,直接加入揮發(fā)油易損失,難以保證產生預期的療效。將揮發(fā)油制成一定的劑型后,再進行二次制劑可以有效增加揮發(fā)油的穩(wěn)定性,使其更好的發(fā)揮療效。目前,常用的揮發(fā)油新劑型有微乳[12]、微球[13]、微囊[14]、脂質體[15]、聚合物膠 束[16]、包合物[17]等。故本研究擬采用羥丙基-β-環(huán)糊精(2-hydroxypropyl-β-cyclodextrin,HPCD)包合技術固化XJP揮發(fā)油,以期增加其穩(wěn)定性,為后續(xù)二次制劑提供保障。
1 儀器與材料
1.1 儀器
Aglient 7820A氣相色譜儀,美國安捷倫公司;DSC-300差示熱量掃描儀,南京大展檢測儀器有限公司;綜合藥品穩(wěn)定性試驗箱,上海一恒科技有限公司;Phenom Pro掃描電子顯微鏡(SEM),Thermo Scientific公司;X射線衍射儀,日本理學XRD Rigaku Ultima IV;Agilent 7890a-5975c氣質聯(lián)用儀,美國Agilent公司。
1.2 材料
蜘蛛香(產地云南大理,批號202109)、木香(產地云南大理,批號202109)、草果(產地云南文山,批號202109),所有藥材經云南中醫(yī)藥大學張潔副教授鑒定,蜘蛛香為敗醬科纈草屬植物蜘蛛香Jones的干燥根莖和根、木香為菊科云木香屬植物木香Decne的干燥根、姜科砂仁屬植物草果Crevost et Lemaire的干燥成熟果實;1,8-桉葉素對照品,中國食品藥品檢定研究院,批號110788-202108,質量分數(shù)99.4%;HPCD,上海源葉生物科技有限公司,批號O11GS163286;無水乙醇,天津市致遠化學試劑有限公司,批號20211001321;溴化鉀,天津市光復精細化工研究所,批號20210316;硅膠G板,青島海洋化工有限公司,批號20201207;香草醛,北京索萊寶科技有限公司,批號1129M011。
2 方法與結果
2.1 揮發(fā)油的提取
按照《中國藥典》2020年版中揮發(fā)油測定甲法提取,依據(jù)前期研究,確定揮發(fā)油提取工藝為稱取500 g蜘蛛香、200 g草果、140 g木香粗粉,加入8倍量的水,浸泡2 h,提取時間5 h,靜置1 h,加入過量無水硫酸鈉,6000 r/min離心(離心半徑4.0 cm)10 min,即得,揮發(fā)油得率(揮發(fā)油得率=所得揮發(fā)油體積/所用藥材質量)為0.60%,每次提取后于4 ℃混合儲存?zhèn)溆茫瑢嶒炃疤崛∽懔繐]發(fā)油使用。
2.2 揮發(fā)油回收率、包合率、收率、含油率的測定
2.2.1 揮發(fā)油回收率測定 取1 mL XJP揮發(fā)油,置于1 L圓底燒瓶中,加入400 mL水,按照《中國藥典》2020年版中揮發(fā)油測定甲法測定,保持微沸3 h,靜置1 h,讀數(shù),即得空白回收率(空白回收率=回收量/加入量)。結果顯示,揮發(fā)油平均空白回收率為82.33%,RSD值為1.83%(=6)。
取1 mL XJP揮發(fā)油,置于1 L圓底燒瓶中,加入10 g HPCD及400 mL水,其余操作同空白回收率測定,讀數(shù),即得加入HPCD后的回收率(加入HPCD后揮發(fā)油空白回收率=回收量/加入量)。結果顯示,揮發(fā)油平均空白回收率為80.67%,RSD值為0.92%(=6)。
2.2.2 包合率測定 取包合物適量,置于1 L圓底燒瓶中,加入400 mL水,其余步驟同“2.2.1”項揮發(fā)油回收率測定,讀取提取出的揮發(fā)油量,計算包合率,計算公式如下。
揮發(fā)油包合率=包合物中揮發(fā)油提取量/(揮發(fā)油加入量×加入HPCD后的揮發(fā)油回收率)
2.2.3 包合物收率測定 包合物制備完成后計算包合物收率,計算公式如下。
包合物收率=包合物質量/(HPCD質量+揮發(fā)油質量)
2.2.4 包合物含油率測定 包合物制備完成后計算包合物含油率,計算公式如下。
包合物含油率=包合物中揮發(fā)油質量/包合物質量
2.2.5 綜合評分() 參照文獻方法[18]計算。
=(1/1max×0.6+2/2max×0.2+3/3max×0.2)×100
1、2、3分別為包合率、收率及含油率,1max、2max及3max分別為最大包合率、最大收率及最大含油率
2.3 XJP揮發(fā)油包合物制備方法考察
2.3.1 單相溶液法 稱取10 g HPCD至50 mL燒杯中,加入20 mL無水乙醇,于45 ℃下攪拌溶解,后逐滴加入1 mL XJP揮發(fā)油(質量0.91 g,與無水乙醇1∶1混勻),保持45 ℃,在900 r/min轉速下攪拌2 h,40 ℃減壓濃縮干燥,石油醚洗滌3次,室溫下?lián)]至恒定質量,即得XJP揮發(fā)油包合物。
2.3.2 飽和水溶液法 稱取10 g HPCD至50 mL燒杯中,加入20 mL純凈水,于45 ℃下攪拌溶解,后逐滴加入1 mL XJP揮發(fā)油(與無水乙醇1∶1混勻),保持45 ℃,在900 r/min轉速下攪拌2 h,50 ℃減壓濃縮干燥,石油醚洗滌3次,室溫下?lián)]至恒定質量,即得XJP揮發(fā)油包合物。
2.3.3 研磨法 稱取10 g HPCD至研缽中,加入2.5 mL純凈水,研磨至糊狀,逐滴加入1 mL XJP揮發(fā)油(與無水乙醇1∶1混勻),繼續(xù)研磨45 min,45 ℃下烘干,石油醚洗滌3次,室溫下?lián)]至恒定質量,即得XJP揮發(fā)油包合物。
2.3.4 超聲法 稱取10 g HPCD至50 mL燒杯中,加入20 mL純凈水,超聲溶解,逐滴加入1 mL XJP揮發(fā)油(與無水乙醇1∶1混勻),超聲(功率100 W)1 h,50 ℃減壓濃縮干燥,石油醚洗滌3次,室溫下?lián)]至恒定質量,即得XJP揮發(fā)油包合物。
4種包合方法比較結果見表1。
2.4 單因素實驗考察XJP揮發(fā)油包合物制備工藝
通過文獻查閱[19-20],選擇HPCD與揮發(fā)油的質量體積比、HPCD與無水乙醇的質量體積比、包合溫度、包合時間以及攪拌轉速作為影響因素進行單因素實驗,單因素實驗設計及結果見表2~6。
2.5 XJP揮發(fā)油包合物中1,8-桉葉素含量測定方法的建立
2.5.1 色譜條件 毛細管柱Agilent 19091J-413 HP-5(30 m×320 μm×0.25 μm),F(xiàn)ID檢測器;進樣口溫度200 ℃,檢測器溫度250 ℃,柱溫以60 ℃為起始溫度,保持4 min,以5 ℃/min的速率升溫至100 ℃,不分流,進樣量1 μL。
表1 4種包合方法的比較(, n = 3)
Table 1 Comparison of four inclusion methods (, n = 3)
包合方法包合率/%收率/%含油率/%綜合評分 單相溶液法82.23±0.7293.37±0.557.35±0.0884.42±0.57 飽和水溶液法26.86±1.8981.55±8.762.75±0.3238.54±2.24 研磨法51.66±1.2481.00±3.635.32±0.0658.64±1.48 超聲法39.67±2.5881.39±12.074.07±0.6748.94±3.15
表2 HPCD與無水乙醇的質量體積比對綜合評分的影響 (, n = 3)
Table 2 Effect of (hydroxypropyl-β-cyclodextrin)-(absolute ethanol) ratio on comprehensive score (, n = 3)
HPCD與無水乙醇的質量體積比包合率/%收率/%含油率/%綜合評分 4∶2067.15±1.7987.08±6.246.43±0.4371.79±4.50 6∶2077.82±2.3981.37±12.897.98±1.2379.96±2.46 8∶2075.93±1.5593.53±3.276.77±0.3370.43±1.14 10∶2082.23±0.7293.37±0.557.35±0.0884.42±0.57 12∶2074.03±2.6082.11±7.237.52±0.4476.93±2.59
表3 HPCD與揮發(fā)油的質量體積比對綜合評分的影響(, n = 3)
Table 3 Effect of (hydroxypropyl-β-cyclodextrin)-(volatile oils) ratio on comprehensive score (, n = 3)
HPCD與揮發(fā)油的質量體積比包合率/%收率/%含油率/%綜合評分 4∶163.14±2.5093.74±4.4412.48±0.9979.36±2.39 7∶173.74±1.5088.23±1.029.62±0.2081.06±1.30 10∶182.23±0.7293.37±0.557.35±0.0884.42±0.57 13∶181.14±0.9387.85±2.736.04±0.1380.65±0.99 16∶179.34±1.9890.80±3.654.70±0.2978.10±1.25
表4 包合溫度對綜合評分的影響(, n = 3)
Table 4 Effect of inclusion temperature on comprehensive score (, n = 3)
包合溫度/℃包合率/%收率/%含油率/%綜合評分 2577.27±1.8989.83±2.767.18±0.3480.15±1.42 3578.10±1.2493.61±1.136.96±0.1981.16±0.88 4582.23±0.7293.37±0.557.35±0.0884.42±0.57 5587.19±3.1290.86±5.798.00±0.3088.16±2.86 6572.31±1.8981.18±1.857.43±0.2975.74±1.52
表5 包合時間對綜合評分的影響(, n = 3)
Table 5 Effect of inclusion time on comprehensive score (, n = 3)
包合時間/h包合率/%收率/%含油率/%綜合評分 0.573.14±1.2488.79±0.456.87±0.0976.76±1.03 1.075.62±1.2492.64±2.076.81±0.2679.10±0.79 2.082.23±0.7293.37±0.557.35±0.0884.42±0.57 3.074.79±2.5891.23±3.816.84±0.0578.30±2.42 4.073.25±1.4384.66±1.247.22±0.2176.49±1.11
表6 攪拌轉速對綜合評分的影響(, n = 3)
Table 6 Effect of stirring speed on comprehensive score(, n = 3)
攪拌轉速/(r?min?1)包合率/%收率/%含油率/%綜合評分 50079.34±1.2489.28±2.827.41±0.3081.76±0.92 70076.03±1.8992.12±1.896.88±0.0379.38±1.69 90082.23±0.7293.37±0.557.35±0.0884.42±0.57 110076.44±1.8991.93±1.306.94±0.2379.69±1.46 130076.86±1.2491.38±4.497.01±0.2779.96±1.33
2.5.2 對照品溶液的制備 取1,8-桉葉素對照品適量,加入適量無水乙醇將其配制成質量濃度分別為814.5、407.3、203.4、162.9、81.5、40.7、16.3 μg/mL的對照品溶液。
2.5.3 供試品溶液的制備 取0.5 g包合物,加入20 mL無水乙醇,超聲30 min后,補足,過0.22 μm濾頭,即得供試品溶液。
2.5.4 專屬性考察 分別取1,8-桉葉素對照品溶液、供試品溶液和空白供試品溶液(取0.5 g HPCD加入20 mL無水乙醇,超聲30 min后,補足,過0.22 μm濾頭,即得空白供試品溶液)進行專屬性考察。結果顯示供試品溶液中1,8-桉葉素峰形良好,HPCD對其檢測無干擾,表明該方法測定包合物中的1,8-桉葉素準確可行,具體結果見圖1。
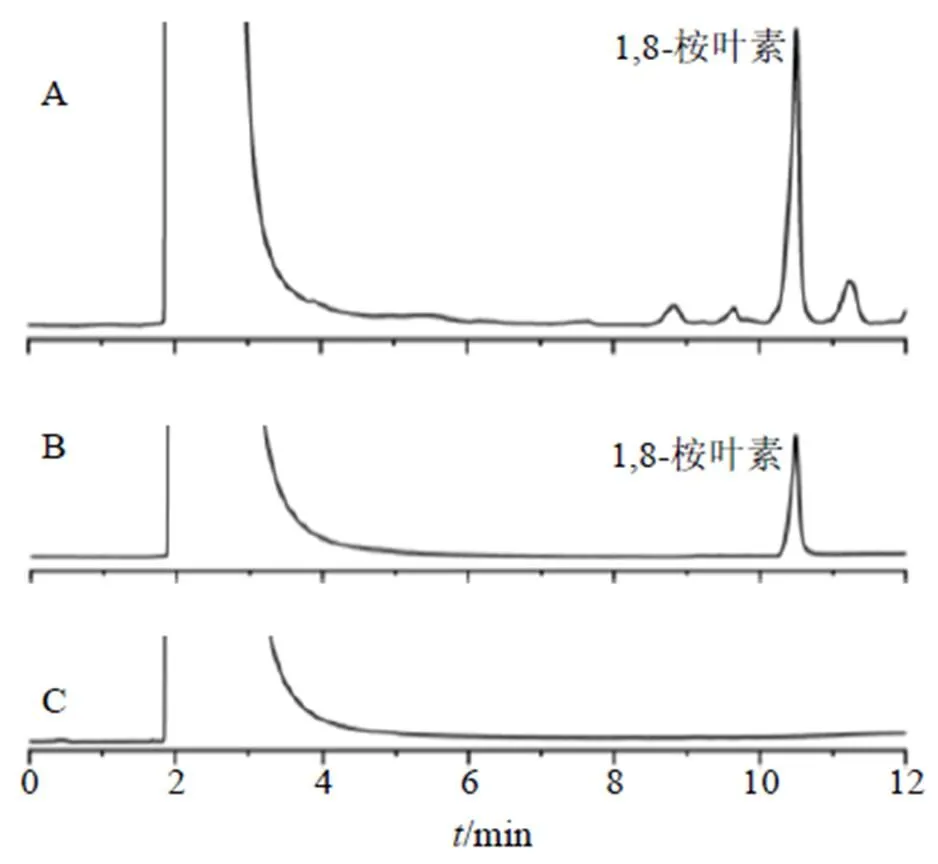
圖1 供試品溶液(A)、1,8-桉葉素對照品溶液(B)和空白供試品溶液(C)的GC圖
2.5.5 線性關系考察 取“2.5.2”項下配制的對照品溶液,按“2.5.1”項色譜條件進樣測定,結果顯示1,8-桉葉素在16.3~814.5 μg/mL線性關系良好,線性方程為=2 876.680 7-2.168 5,=1.000 0。
2.5.6 精密度考察 取1,8-桉葉素對照品溶液,過0.22 μm濾膜后,按“2.5.1”項色譜條件連續(xù)進樣6針,結果顯示,1,8-桉葉素峰面積的RSD為1.72%,表明儀器精密度良好。
2.5.7 重復性考察 平行制備6份供試品溶液,按“2.5.1”項色譜條件進樣,結果顯示XJP揮發(fā)油包合物中1,8-桉葉素質量分數(shù)的RSD為0.86%,表明該方法重復性良好。
2.5.8 穩(wěn)定性考察 取供試品溶液,分別于制備后0、1、2、4、6、8、12、24 h按“2.5.1”項色譜條件進樣,結果顯示1,8-桉葉素峰面積的RSD為1.83%,表明供試品溶液在24 h內穩(wěn)定性良好。
2.5.9 加樣回收率考察 取包合物9份,分別加入低、中、高質量濃度的1,8-桉葉素對照品溶液各3份,按照“2.5.3”項方法處理后,按“2.5.1”項色譜條件進樣測定,計算1,8-桉葉素加樣回收率。結果顯示1,8-桉葉素高、中、低質量濃度平均回收率分別為99.43%、95.98%、98.12%,RSD分別為1.71%、0.57%、0.57%。
2.6 正交試驗優(yōu)化XJP揮發(fā)油包合物制備工藝
2.6.1 正交試驗因素、水平的確定 參照單因素實驗結果,可知HPCD與揮發(fā)油的質量體積比、攪拌轉速對試驗的影響較小,且當HPCD與揮發(fā)油的質量體積比為10∶1,攪拌轉速為900 r/min時綜合評分最高,故固定二者。選擇HPCD與無水乙醇的質量體積比(1)、包合溫度(2)以及包合時間(3)作為影響因素,以包合率(1)、收率(2)、含油率(3)及包合物中1,8-桉葉素含量(4)為指標,建立L9(34)正交試驗,結果見表7。
表7 正交試驗因素水平及結果
Table 7 Orthogonal experimental factor-levels and results
編號X1X2/℃X3/hX4(誤差)Y1/%Y2/%Y3/%Y4/(mg?g?1)綜合評分 18∶20 (1)45 (1)1 (1)(1)75.9397.966.5217.16 90.79 28∶20 (1)55 (2)2 (2)(2)80.5896.247.0419.39 94.84 38∶20 (1)65 (3)3 (3)(3)74.3891.776.8218.26 89.40 410∶20 (2)45 (1)2 (2)(3)76.8694.506.8417.85 91.07 510∶20 (2)55 (2)3 (3)(1)80.5895.607.0919.50 94.79 610∶20 (2)65 (3)1 (1)(2)76.8698.266.5817.03 91.28 712∶20 (3)45 (1)3 (3)(2)82.6494.717.3420.10 96.24 812∶20 (3)55 (2)1 (1)(3)86.7795.337.6620.45 98.97 912∶20 (3)65 (3)2 (2)(1)82.6491.667.5819.79 95.30 K1275.03278.10281.04280.88 K2277.14288.60281.21282.36 K3290.51275.98280.43279.44 R15.4812.620.782.92
2.6.2 信息熵權法確定各指標權重系數(shù)[17]
(1)建立原始數(shù)據(jù)矩陣:現(xiàn)有個待評項目,個評價指標,建立原始數(shù)據(jù)矩陣=(A)×n,其中A為第個指標下第個項目的評價值,為正交試驗次數(shù)(=9),為評價指標(=4)。
(2)原始數(shù)據(jù)矩陣標準化處理:由于評價指標單位不統(tǒng)一,故在數(shù)據(jù)處理前需進行指標標準化處理,將原始數(shù)據(jù)矩陣=(A)×n轉化為標準化數(shù)據(jù)矩陣=(X)×n。正交試驗中包合率、收率、含油率及包合物中1,8-桉葉素含量均為正向指標,標準化公式為X=(A-Amin)/(Amax-Amin),其中X為標準化數(shù)據(jù)。
(3)標準化數(shù)據(jù)矩陣歸一化處理:進行指標歸一化處理,將標準化數(shù)據(jù)矩陣=(X)×n轉化為標準化數(shù)據(jù)概率矩陣=(P)×n。轉化公式為P=X/X,其中P表示第個指標下第個項目的概率,且0≤P≤1。
(4)計算指標信息熵(H):計算第個指標的H,計算公式如下。
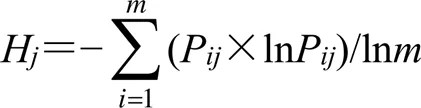
當P=0時,lnP無意義,故P=0時,將P×lnP修正為0。
包合率、收率、含油率及包合物中1,8-桉葉素含量的信息熵H分別為0.866 6、0.871 8、0.953 5、0.936 2。
(5)計算各指標的差異系數(shù)(g)及熵權系數(shù)(W):g計算公式為g=1-H;W計算公式為W=g/g,其中0≤W≤1。
包合率、收率、含油率及包合物中1,8-桉葉素含量的g分別為0.133 4、0.128 2、0.046 5、0.063 8,W分別為0.358 6、0.344 7、0.125 1、0.171 5。
(6)綜合評分的計算:綜合評分=(1/1max×0.358 6+2/2max×0.344 7+3/3max×0.125 1+4/4max×0.171 5)×100。
2.6.3 結果分析 由表8方差分析結果可知,各因素對綜合評分的影響大小為1>2>3,其中1、2對綜合評分有顯著性的影響(<0.05),最佳工藝為HPCD與揮發(fā)油的質量體積比為10∶1,HPCD與無水乙醇的質量體積比為12∶20,包合溫度55 ℃,包合時間2 h,攪拌轉速900 r/min。
表8 方差分析
Table 8 Analysis table of variance
因素偏差平方和自由度均方F值顯著性 X146.982 2223.491 133.058 1P<0.05 X230.445 4215.222 721.422 3P<0.05 X30.112 220.056 10.078 9 X4 (誤差)1.421 220.710 61.000 0
0.05(2, 2)=19.00
2.6.4 驗證實驗 按照“2.6.3”項中確定的最佳工藝,平行制備3份XJP揮發(fā)油包合物,具體工藝為稱取12 g HPCD至50 mL燒杯中,加入20 mL無水乙醇,于55 ℃下攪拌溶解,后逐滴加入1.2 mL XJP揮發(fā)油(與無水乙醇1∶1混勻),保持55 ℃,在900 r/min的轉速下攪拌2 h,40 ℃減壓濃縮干燥,石油醚洗滌3次,室溫下?lián)]至恒定質量,即得。實驗結果見表9,制得的包合物平均包合率為88.15%,收率為96.11%,含油率為7.65%,包合物中1,8-桉葉素的含量為20.28 mg/g,綜合評分為99.67,RSD值為0.36%,表明該工藝穩(wěn)定。
表9 驗證實驗結果
Table 9 Results of verification test
編號包合率/%收率/%含油率/%1,8-桉葉素/(mg?g?1)綜合評分RSD/% 187.8194.497.7520.4599.260.36 287.8197.857.4920.3399.91 388.8496.017.7220.0699.84 平均值88.1596.117.6520.2899.67
2.7 包合物的表征
2.7.1 薄層色譜(TLC)分析 對HPCD、1,8-桉葉素、揮發(fā)油、包合后的揮發(fā)油、包合物及物理混合物(1.2 mL揮發(fā)油加入12 g HPCD中研磨20 min)進行TLC分析,其中HPCD、包合物及物理混合物按照“2.5.3”項方法進行處理,1,8-桉葉素、揮發(fā)油、包合后的揮發(fā)油則取適量無水乙醇稀釋。吸取上述樣品溶液,分別點樣于同一硅膠G薄層板上,以正己烷-醋酸乙酯(17∶3)為展開劑,展開,取出,晾干,噴以5%香草醛硫酸溶液,于105 ℃下烘烤至斑點顯色清晰。結果見圖2,可見HPCD未顯示出斑點,揮發(fā)油、包合后的揮發(fā)油、包合物及物理混合物中化學成分幾乎一致,均含有1,8-桉葉素,表明包合前后揮發(fā)油中主要成分未發(fā)生變化。
2.7.2 紫外吸收光譜 對HPCD、揮發(fā)油、包合物及物理混合物進行紫外吸收光譜分析,取HPCD、包合物、物理混合物、揮發(fā)油適量無水乙醇溶解。將上述樣品進行紫外吸收光譜分析,掃描范圍200~400 nm。結果見圖3,揮發(fā)油、物理混合物在300 nm處有紫外吸收,而包合物、HPCD中未見紫外吸收,且二者吸收曲線相似,表明包合物已形成。

1-HPCD 2-1,8-桉葉素 3-揮發(fā)油 4-包合后的揮發(fā)油 5-包合物 6-物理混合物
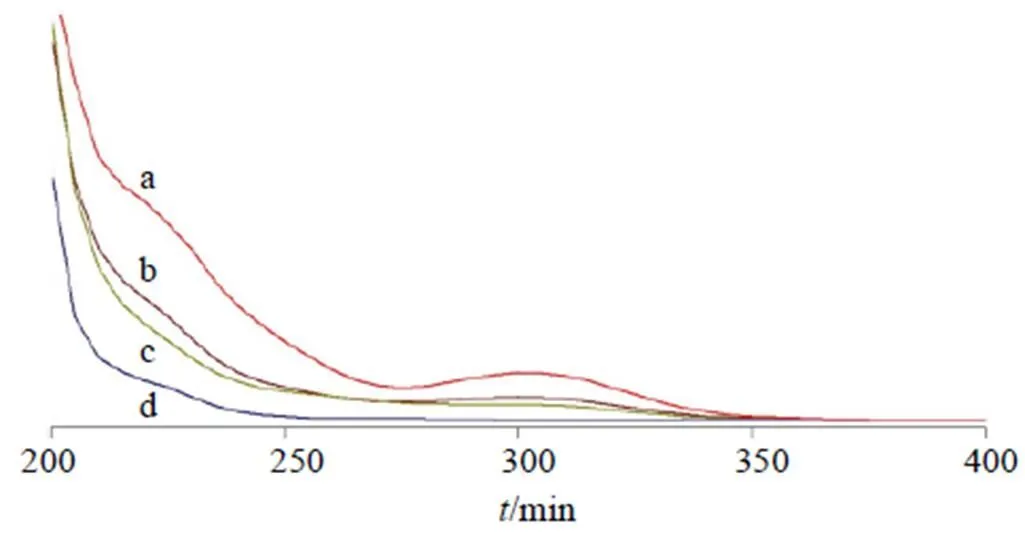
a-揮發(fā)油 b-物理混合物 c-包合物 d-HPCD
2.7.3 傅里葉紅外光譜(FTIR)分析 取HPCD、物理混合物、包合物及揮發(fā)油適量,將之與溴化鉀共同壓片,在4000~400 cm?1波數(shù)進行FTIR分析。結果見圖4,可知揮發(fā)油在3 453.51、2 926.84、1 677.87 cm?1有較強的特征吸收峰,且在指紋區(qū)1 155.53~669.20 cm?1存在多個吸收峰,而包合物中2 929.62、1 669.48 cm?1處吸收峰明顯減弱,表明揮發(fā)油已經進入HPCD空腔中,導致其紅外震動受限。HPCD、物理混合物以及包合物3者紅外吸收圖譜相似,但又存在細微差別,如HPCD在2 362.42 cm?1處表現(xiàn)有特征吸收,但物理混合物、包合物中此處吸收峰消失;物理混合物、包合物分別在2 925.43、2 929.62 cm?1及1 673.67、1 669.48 cm?1處存在特征吸收峰,且峰形、峰面積存在差異,而兩者指紋區(qū)圖譜也存在一定差別,表明二者為不同物象。
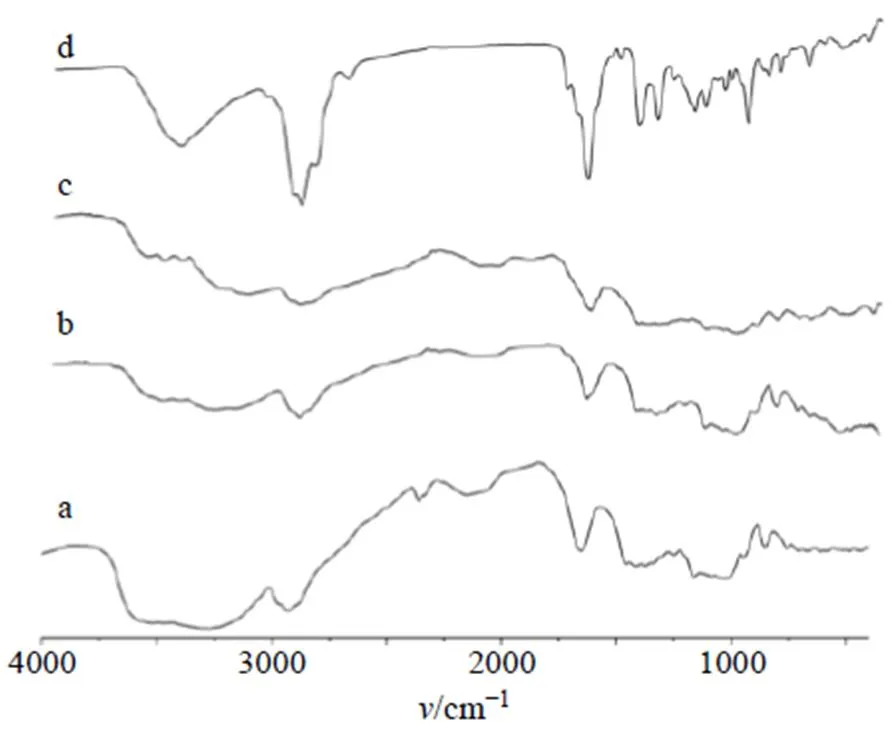
a-HPCD b-物理混合物 c-包合物 d-揮發(fā)油
2.7.4 差示掃描量熱法(differential scanning calorimetry,DSC)分析 對HPCD、揮發(fā)油物理混合物、揮發(fā)油、包合物進行DSC分析,掃描條件為以空氧化鋁(Al2O3)坩堝為空白參比,另一坩堝中加入待測物質,掃描范圍為25~400 ℃,升溫程序為10 ℃/min,測定氣體為空氣。結果見圖5,HPCD、揮發(fā)油分別在56.5、49.0 ℃處有特征吸熱峰,物理混合物的特征吸熱峰表現(xiàn)在55.3 ℃處,此處特征吸熱峰的改變可能是由于揮發(fā)油中一些低沸點物質的存在,使得物理混合物的特征吸熱峰降低。而包合物則在83.4 ℃表現(xiàn)出1個特征吸熱峰,且未表現(xiàn)揮發(fā)油在141.6 ℃處的特征吸熱峰,且其熱熵值變化明顯,表明形成了新的物相,且其熱穩(wěn)定性有著一定的提升。此外,包合物除在326.4~364.1 ℃處與HPCD存在一定差異外,其余溫度均表現(xiàn)出與HPCD相似的DSC圖,也表明包合物已形成。

a-HPCD b-物理混合物 c-包合物 d-揮發(fā)油
2.7.5 SEM觀察 取適量HPCD、物理混合物、包合物,真空鍍金后固定于銅板上,分別于500×、5000×倍數(shù)下通過SEM觀測各樣品形貌。結果見圖6,HPCD為扁平空心球狀結構,且表面凹凸不平,分布有細小孔洞。物理混合物仍表現(xiàn)出與HPCD相似的形貌,但體積較HPCD更小,推測可能是由于研磨導致原有扁平空心球狀結構破壞,但破壞不完全。包合物則表現(xiàn)出與HPCD、物理混合物完全不同的形貌,原有扁平空心球狀結構消失,變?yōu)楸砻孑^為平整、光滑的塊狀結構。
2.7.6 X射線衍射(XRD)分析 取HPCD、物理混合物、包合物進行XRD檢測,實驗條件為Cu靶(電壓40 kV,電流40 mA),步進掃描0.02°/步,掃描范圍10°~80°,掃描速度2°/min。結果見圖7,HPCD、物理混合物、包合物均在14°~22.5°有衍射峰,但包合物衍射峰較強,3者XRD圖譜相似,均表現(xiàn)為無定形結構;包合物在22.5°~27°表現(xiàn)出與HPCD不同的峰形。表明包合揮發(fā)油后,其晶型結構并未發(fā)生顯著改變。

圖6 樣品掃描電鏡結果
2.7.7 揮發(fā)油包合前后成分變化 分別取包合前、后揮發(fā)油100 μL,加入無水乙醇稀釋至5 mL,搖勻,過0.22 μm濾頭,按下述條件檢測,檢測結果見圖8。使用中南大學中藥現(xiàn)代化研究中心研發(fā)的“中藥指紋圖譜計算軟件”,以平均圖譜為標準計算相似度,包合前、后揮發(fā)油GC-MS指紋圖譜相似度為0.979、0.979,表明包合前后揮發(fā)油相似,而包合前、后某些成分存在差異可能是由于提取包合物中揮發(fā)油的過程所致。
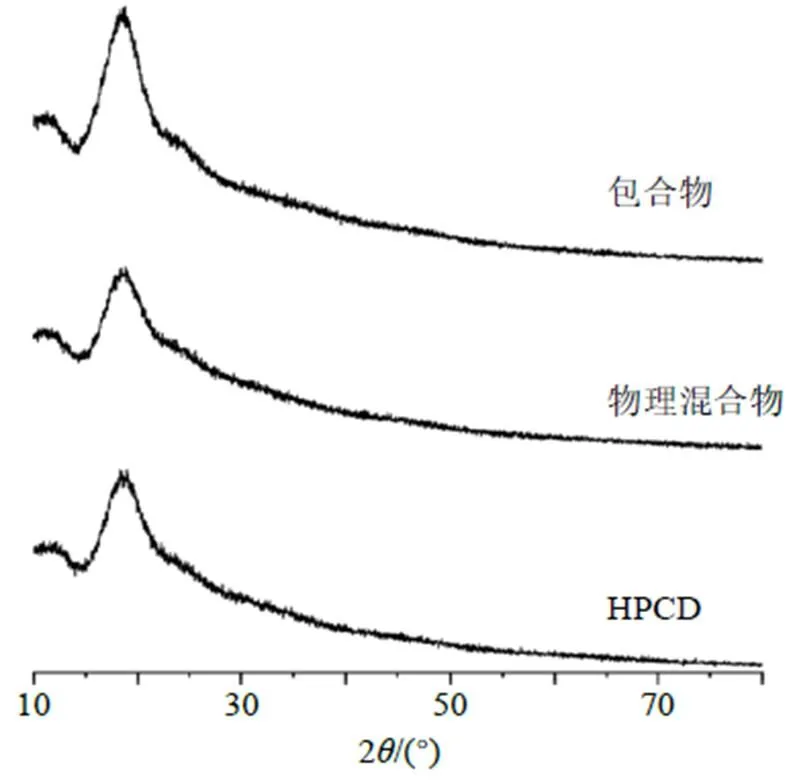
圖7 樣品X射線衍射結果
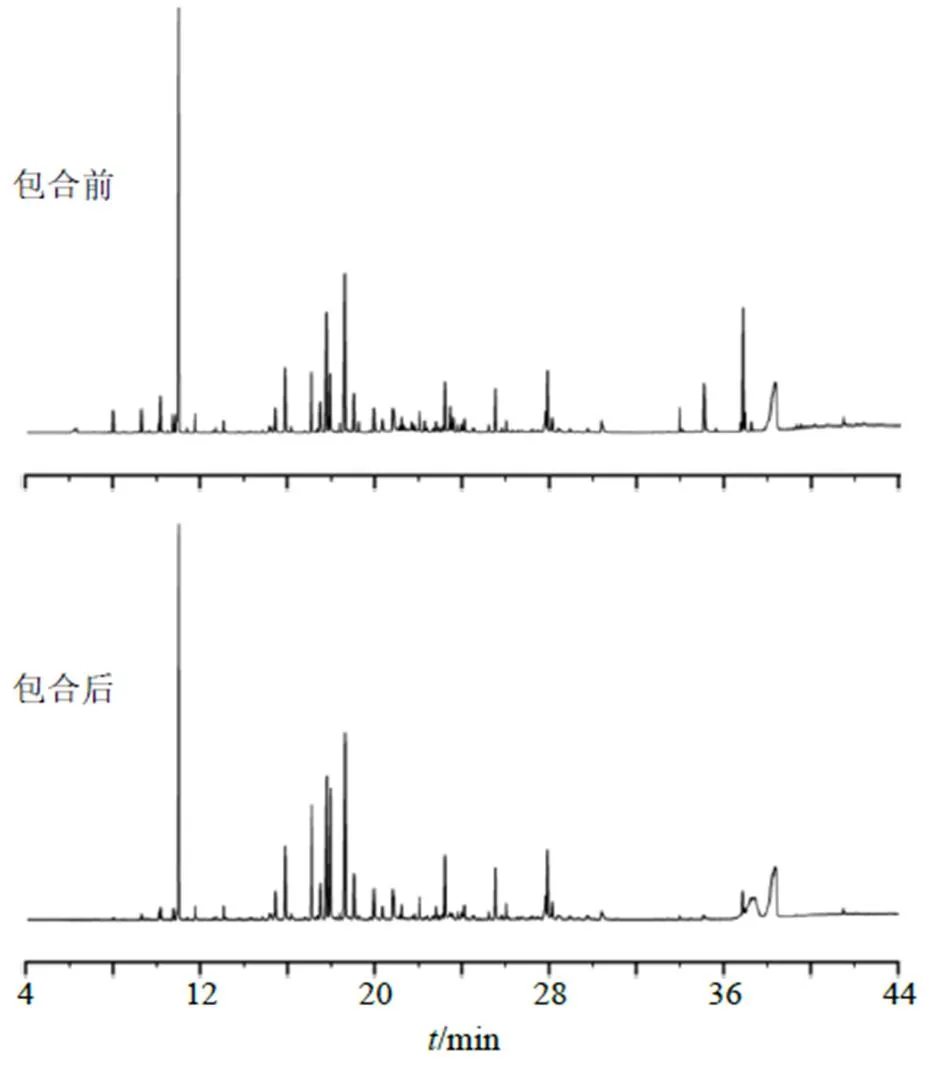
圖8 包合前、后揮發(fā)油GC-MS結果
GC條件:J&W 122-5532石英毛細管柱(30 m×250 μm×0.25 μm),柱溫40 ℃(保持1 min),以5 ℃/min的速率升溫至180 ℃(保持2 min),以8 ℃/min的速率升溫至250 ℃(保持5 min),進樣口溫度230 ℃,載氣為氦氣,體積流量1 mL/min;分流比40∶1,進樣量1 μL,溶劑延遲4 min。
質譜條件:電離方式EI,離子源230 ℃,電子能量70 eV,四極桿溫度150 ℃,輔助加熱290 ℃,掃描質量范圍/50~600,質譜檢索標準庫:NIST 11。
2.7.8 包合物穩(wěn)定性影響因素考察
(1)高溫試驗:精密稱取3份包合物,每份1.0 g,置于燒杯中,放置于藥品穩(wěn)定性試驗箱,設定溫度為60 ℃,于第0、5、10天取樣,觀察包合物外觀(圖9)及氣味,并測定包合物中的1,8-桉葉素的含量,計算1,8-桉葉素損失率。包合物在第5天時,外觀為白色粉末,無揮發(fā)油氣味;在第10天時,包合物泛黃,有輕微揮發(fā)油氣味。在第10天時,每克包合物中1,8-桉葉素損失率為29.55%,結果提示包合物儲存時應避免高溫,具體結果見表10。
(2)高濕試驗:精密稱取3份包合物,每份1.0 g,置于燒杯中,放置于藥品穩(wěn)定性試驗箱,設定濕度為90%,于第0、5、10天取樣,觀察包合物外觀(圖9)及氣味,并測定包合物中的1,8-桉葉素的含量,計算1,8-桉葉素損失率。在第5天時,包合物結聚為透明塊狀物,在第10天時,包合物結聚為淡黃色透明塊狀物;在第5、10天均無揮發(fā)油氣味。在第10天時,包合物中1,8-桉葉素損失率為31.75%,結果提示包合物應當儲存于干燥處,具體結果見表11。

圖9 包合物外觀
表10 高溫試驗
Table 10 High temperature test
t/d1,8-桉葉素/(mg?g?1)損失率/% 123平均值 020.5920.5920.5920.590 515.9516.1316.1516.0821.88 1014.2414.5814.6914.5029.55
(3)強光試驗:精密稱取3份包合物,每份1.0 g,置于燒杯中,放置于藥品穩(wěn)定性試驗箱,設定光強為4500 lx,于第0、5、10天取樣,觀察包合物外觀(圖9)及氣味,并測定包合物中的1,8-桉葉素的含量,計算1,8-桉葉素損失率。在第10天時,包合物仍為白色粉末,無揮發(fā)油氣味。在第10天時,包合物中1,8-桉葉素損失率為21.80%,結果提示光照對包合物儲存的影響較小,但仍應避光處理,具體結果見表12。
表11 高濕試驗
Table 11 High humidity test
t/d1,8-桉葉素/(mg?g?1)損失率/% 123平均值 020.5920.5920.5920.590 516.4216.0816.3716.2920.86 1014.0414.4013.7114.0531.75
表12 強光試驗
Table 12 Strong light test
t/d1,8-桉葉素/(mg?g?1)損失率/% 123平均值 020.5920.5920.5920.590 517.0517.4917.0417.1916.84 1016.0816.1416.0716.1021.80
3 討論
XJP善治因氣機壅遏、氣滯不通所致小兒腹痛。臨床使用往往忽略了其揮發(fā)油類成分,而研究發(fā)現(xiàn)蜘蛛香、木香、草果揮發(fā)油均具有一定的藥理作用。但揮發(fā)油理化性質不穩(wěn)定,易分解,通常將其制成一定的劑型來提高其穩(wěn)定性。包合物是指將藥物包載于環(huán)糊精空腔內,顯著提升藥物穩(wěn)定性的一種給藥形式。目前包合工藝已經較為成熟,有飽和水溶液法[21]、研磨法[22]、超聲法[20]、膠體磨法[21]、單相溶液法[20]等。單相溶液法是指以無水乙醇溶解HPCD及揮發(fā)油,形成單相溶液,制備包合物的一種方法。其包合率顯著高于飽和水溶液法、研磨法、超聲法,可能與增加了揮發(fā)油和HPCD分子間接觸幾率有關[20]。
信息熵權法是一種客觀賦權方法,其原理是依據(jù)指標評價結果,根據(jù)各指標的變異程度所反映的信息量來確定各指標權重,相較多指標評價通常采用的主觀賦權法,信息熵權法能有效避免賦權時的主觀性,更客觀的對信息結果進行評價[17, 23-24]。本研究發(fā)現(xiàn),基于信息熵權法優(yōu)選單相溶液法制備XJP揮發(fā)油HPCD包合物最佳工藝為:稱取12 g HPCD至50 mL燒杯中,加入20 mL無水乙醇,于55 ℃下攪拌溶解,后逐滴加入1.2 mL XJP揮發(fā)油(與無水乙醇1∶1混勻),保持55 ℃,在900 r/min的轉速下攪拌2 h,40 ℃減壓濃縮干燥,石油醚洗滌3次,室溫下?lián)]至恒定質量。對制得的包合物進行多種表征,驗證了包合物的形成。影響因素試驗表明,包合物應當避光、低溫、低濕保存。該工藝簡便、穩(wěn)定,且使用的儀器較少,適合后續(xù)工廠放大生產,為后續(xù)進一步研究及產品開發(fā)提供依據(jù)。
利益沖突 所有作者均聲明不存在利益沖突
[1] 黃寶康, 鄭漢臣, 張巧艷, 等. 纈草和蜘蛛香的資源分布及民族藥用調查 [J]. 中國野生植物資源, 2006, 25(1): 12-15.
[2] Zhang B, Wang Y, Jiang C M,.Jones inhibits Rotavirus-induced diarrhea via phosphatidylinositol 3-kinase/protein kinase B signaling pathway [J]., 2021, 31(8): 1115-1122.
[3] Wang L W, Sun Y, Zhao T T,. Antidepressant effects and mechanisms of the total iridoids ofon the brain-gut axis [J]., 2020, 86(3): 172-179.
[4] Xu Y N, Guo P X, Wang Y L,. Effect and mechanism of ethanol extracts of Muxiang () on gastric ulcers in rats [J]., 2020, 40(1): 59-66.
[5] Zheng H, Chen Y L, Zhang J Z,. Evaluation of protective effects of costunolide and dehydrocostuslactone on ethanol-induced gastric ulcer in mice based on multi-pathway regulation [J]., 2016, 250: 68-77.
[6] 楊偉倩, 田洋, 張愛靜, 等. 草果水提物對洛哌丁胺誘導的小鼠便秘癥狀的影響 [J]. 西南農業(yè)學報, 2020, 33(10): 2209-2214.
[7] 趙兵, 郝萍, 高昂, 等. 纈草與蜘蛛香揮發(fā)油的抗菌抗氧化活性研究 [J]. 天然產物研究與開發(fā), 2013, 25(8): 1037-1040.
[8] 陳飛龍, 譚曉梅, 湯慶發(fā), 等. 不同產地木香揮發(fā)油成分的GC-MS分析比較 [J]. 中國藥房, 2011, 22(23): 2187-2189.
[9] 吳惠妃, 梅全喜, 李慶國, 等. 白木香種子揮發(fā)油化學成分及抗氧化性研究 [J]. 中藥材, 2013, 36(9): 1463-1466.
[10] 唐飛, 劉美辰, 敖慧. 木香與川木香揮發(fā)油化學成分及抗菌活性的對比研究 [J]. 中華中醫(yī)藥學刊, 2020, 38(6): 165-168.
[11] 張琪, 黃燕, 楊揚. 草果揮發(fā)油的研究進展 [J]. 時珍國醫(yī)國藥, 2014, 25(4): 931-933.
[12] 李紹林, 段啟, 趙珍東, 等. 石菖蒲揮發(fā)油微乳鼻噴劑的制備及其質量評價 [J]. 中草藥, 2019, 50(8): 1935-1941.
[13] 張煥煥, 王云, 張振海, 等. 共聚維酮-Soluplus噴霧干燥微球包合肉桂油研究 [J]. 中草藥, 2020, 51(22): 5723-5729.
[14] 穆啟運, 藍濤華, 徐方方, 等. 正交試驗優(yōu)選川芎和細辛揮發(fā)油的β-環(huán)糊精微囊萃取工藝及成分分析 [J]. 中國實驗方劑學雜志, 2014, 20(20): 1-5.
[15] 管詠梅, 劉佳, 張建林, 等. 解郁安神方中4種混合揮發(fā)油脂質體的制備與評價 [J]. 中國中藥雜志, 2019, 44(7): 1363-1370.
[16] 張壯麗, 王紀芬, 汪婉瑩, 等. 魚腥草揮發(fā)油聚合物膠束的制備 [J]. 中成藥, 2019, 41(7): 1490-1495.
[17] 成余勤, 石森林, 來平凡. 基于熵權法和正交設計優(yōu)化加減當歸芍藥散揮發(fā)油的β-環(huán)糊精包合工藝 [J]. 中草藥, 2021, 52(10): 2951-2957.
[18] 王紅芳, 李國川, 陳層層, 等. 利肝隆顆粒組分揮發(fā)油提取、β-環(huán)糊精包合工藝的優(yōu)化 [J]. 中成藥, 2019, 41(6): 1384-1388.
[19] 王琪, 胡佳亮, 張金良, 等. 薄荷-荊芥穗揮發(fā)油羥丙基-β-環(huán)糊精包合物的制備 [J]. 中成藥, 2020, 42(12): 3122-3128.
[20] 陸兆光, 萬琴, 孟瑾, 等. 青蒿揮發(fā)油羥丙基-β-環(huán)糊精包合物的制備及其抗病毒活性分析 [J]. 中國實驗方劑學雜志, 2018, 24(18): 11-15.
[21] 李海亮, 崔小麗, 仝燕, 等. 2種方法制備4種中藥揮發(fā)油β-環(huán)糊精包合物的規(guī)律性探索 [J]. 中國中藥雜志, 2012, 37(7): 908-912.
[22] 徐佳, 黃一平, 王麗, 等. 石丹顆粒中揮發(fā)油包合工藝優(yōu)選 [J]. 中國實驗方劑學雜志, 2015, 21(1): 32-34.
[23] 曾海蓉, 李婷娜, 冉倩, 等. 基于熵權法結合Box-Behnken響應面法優(yōu)化桂枝芍藥知母顆粒復方提取工藝 [J]. 中草藥, 2020, 51(1): 84-90.
[24] 李珍, 喬向東, 楊洋, 等. 熵權法結合灰色關聯(lián)度法評價白芷飲片質量 [J]. 中國現(xiàn)代應用藥學, 2022, 39(1): 61-67.
Optimization of inclusion process of Xiangguo Jianxiao Pian volatile oils based on the information entropy method and its characterization
HAO Jia-xu1, 2, 3, ZHA Li-chun1, 2, 3, FAN Xiao1, 2, 3, CUI Li-li1, 2, 3, GAO Jia-ju1, 2, 3, ZENG Peng-hui1, 2, 3, WU Xu-run1, MA Yun-shu1, 2, 3
1. School of Chinese Materia Medica, Yunnan University of Chinese Medicine, Kunming 650500, China 2. Key Laboratory of External Drug Delivery System and Preparation Technology in University of Yunnan Province, Kunming 650500, China 3. Yunnan Key Laboratory of Dai and Yi Medicines, Kunming 650500, China
To optimize the best process of 2-hydroxypropyl-β-cyclodextrin (HPCD) inclusion Xiangguo Jianxiao Pian (XJP,香果健消片) volatile oil.On the basis of single factor experiments, taking inclusion rate, yield, oil content, and the content of eucalyptol per gram of inclusion compound as indicators, and the ratio of HPCD to absolute ethanol, inclusion temperature, and inclusion time were influencing factors. The information entropy method was used to determine the weight coefficient of each index, then calculate the comprehensive score, the inclusion process conditions were optimized by orthogonal experiment. The inclusion complex was verified by thin layer chromatography, ultraviolet absorption spectroscopy, Fourier infrared spectroscopy, differential scanning calorimetry, scanning electron microscopy, X-ray diffraction, GC-MS, and investigated the factors affecting stability of the inclusion compound.The best inclusion process was that the ratio of HPCD to absolute ethanol was 12:20(g/mL), the inclusion temperature was 55 ℃, and the inclusion time was 2 h. The average inclusion rate of the obtained inclusion compound was 88.15%, the yield was 96.11%, the oil content was 7.65%, and the content of eucalyptol per gram of the inclusion compound was 20.28 mg. The volatile oil successfully entered the HPCD cavity, and the composition of the volatile oil remained basically unchanged before and after the inclusion, and the inclusion compound had good stability.The preferred volatile oil inclusion process is stable and feasible.
Xiangguo Jianxiao Pian; volatile oil; 2-hydroxypropyl-β-cyclodextrin; eucalyptol; information entropy method; orthogonal design; inclusion process
R283.6
A
0253 - 2670(2022)13 - 3962 - 10
10.7501/j.issn.0253-2670.2022.13.009
2022-01-13
國家自然科學基金資助項目(82060723);國家自然科學基金資助項目(82174065)
郝佳旭,男,碩士研究生,研究方向為中藥藥劑學。E-mail: zermattbm@163.com
馬云淑,女,博士,教授,研究方向為中藥藥劑學。Tel: (0871)65918217 E-mail: yunshuma2@126.com
[責任編輯 鄭禮勝]
