Construction and validation of a novel prediction system for detection of overall survival in lung cancer patients
2022-06-27 08:34:44ChengZhongYunLiangQunWangHaoWeiTanYanLiang
World Journal of Clinical Cases 2022年18期
關鍵詞:學生
lNTRODUCTlON
Lung cancer (LC) is one of the most common malignant tumors and one of the leading causes of cancerrelated deaths worldwide. In 2012, the deaths caused by LC were approximately 1.6 million, accounting for 19% of the total global cancer deaths[1,2]. Despite advancement in its treatment, surgery is the primary therapy for patients with non-small-cell lung carcinoma. However, the overall survival rate of LC patients remains low.
Many factors have an aberrant effect on the overall survival of LC patients. The main reason is that patients who are diagnosed with advanced and metastasis LC cannot undergo radical surgery.Therefore, the development of more advanced diagnosis and predictive biomarkers is a promising direction for cancer diagnosis and treatment[3,4].
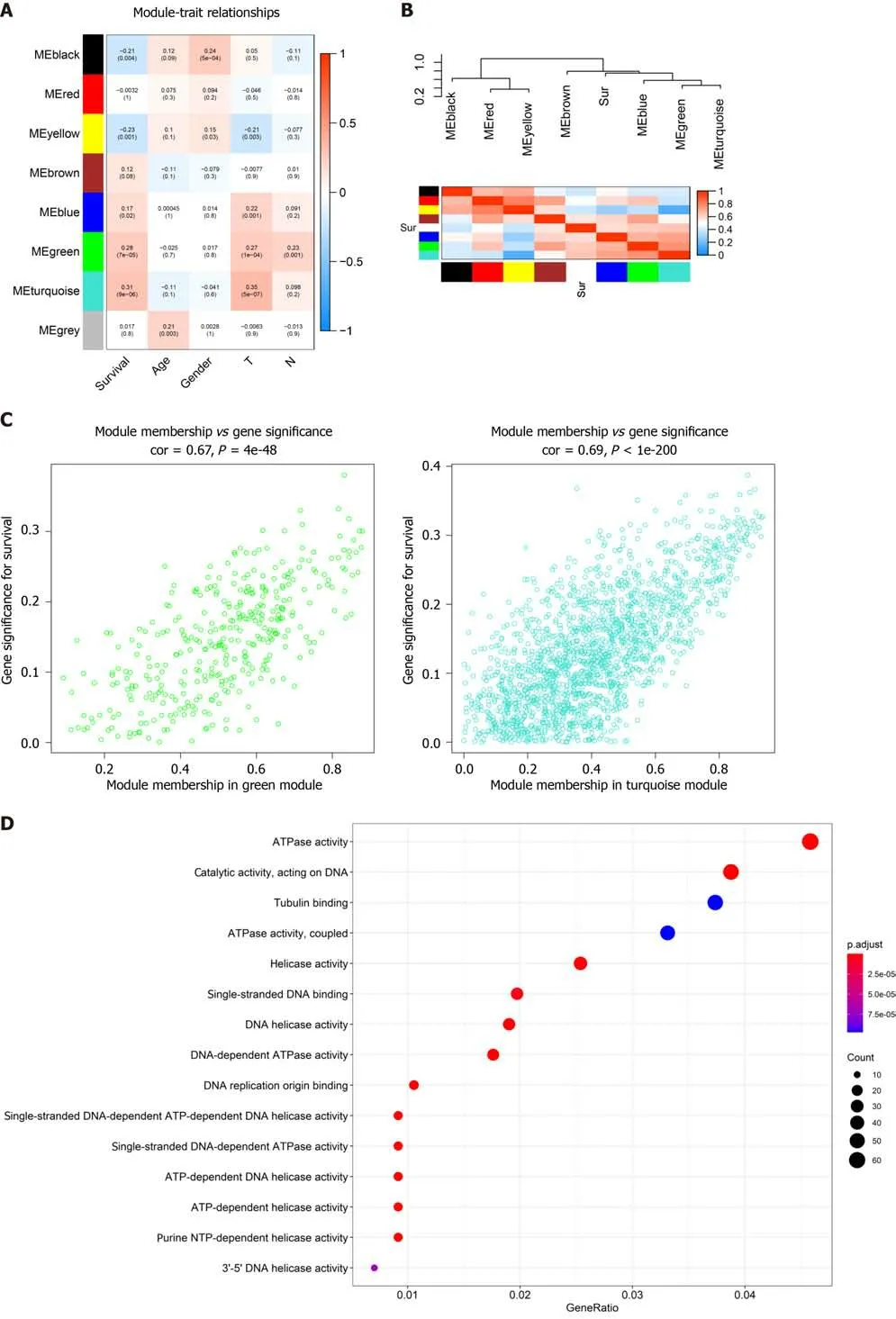
The topological overlap matrix plot (Figure 1G) indicated the correlation between the genes of the eight modules sorted using the clustering tree. The turquoise module showed a positive correlation of about 0.31 with LC patient survival, followed by the green module (0.28) and yellow module (0.23) (Figure 2A and B). The turquoise module contained 1673 genes. We then selected 75 key genes from the turquoise module with MM > 0.8 (Figure 2C). Taken together, the turquoise module was finally selected for further analysis.
In recent years, remarkable progress has been made in immunotherapy, targeted treatment, and promising biomarkers. However, the available treatments and diagnostic methods are not specific for all patients[5]. A high recurrence rate is observed after such treatment because of the complexity of cancer.Identification of new diagnostic and therapeutic biomarkers for cancer treatment is urgent[6].
在他的觀念里,做任何事情都分兩個層次,基礎層和提升層。“一件事情做到某種程度是底線,是必須達到的60分;而從60分到100分,需要不斷提升,是需要一直思考的事情。”
7)是否有政策支持:該指標主要考慮國家政策及發展的方向,例如船舶岸電、電動汽車都有相應國家支持以及指標下達,因此要力度更大才行。
(1)去中心化思想,發行數量固定。法幣的發行受政府與中央銀行約束;但比特幣不同,它采用區塊鏈技術和非對稱密碼技術,發行不受央行約束,而且比特幣的發行具有上限,從而避免一些因為人為決策因素而導致的貨幣貶值。
The identification of differentially expressed genes (DEGs) has garnered considerable scientific attention. However, this method does not consider genes with similar expression patterns. Weighted Gene Co-expression Network Analysis (WGCNA) is a new algorithm that evaluates the correlation between gene modules and clinical features by constructing a scale-free gene coexpression network. In this study, we combined the WGCNA algorithm with DEGs to identify pivotal genes associated with clinicopathological characteristics and to provide insights into targeted therapy of LC.
MATERlALS AND METHODS
Data collection
The clinical and expression data of LC patients were derived from the Gene Expression Omnibus (GEO)and The Cancer Genome Atlas (TCGA) databases (https://portal.gdc.cancer.gov/; http://www.ncbi.nlm.nih.gov/geo/). GEO data contains two cohorts (GSE30129 and GSE50081). The sva package was used to normalize the Meta-GEO data. Next, we used the TCGA data (LogFC > 0.5,
< 0.05) to identify the DEGs and combined these DEGs with all GEO genes. Finally, 5007 genes were selected for the subsequent analyses.
Construction of WGCNA
WGCNA R package was used to analyze the coexpression networks. We determined the threshold of β= 5 to establish the optimal weighted network by Pearson’s correlational analysis. The adjacent matrix was transformed into a topological overlap measure matrix
topological overlapping dissimilarity to estimate its connectivity property in the network. We set the minimum number of module genes to 100,and the threshold for merging similar modules was set to 0.25.
< 0.05 was considered to indicate statistical significance. After the modules of interest were selected, the key genes were selected according to the gene signature (GS) and module membership (MM) of each module.
Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analyses
The functional analysis of core genes was performed using the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). Through the R language clusterProfiler and ggplot2 packages, several important pathways have been discovered so far. The cut-off criteria were defined as count > 2 and
< 0.05.
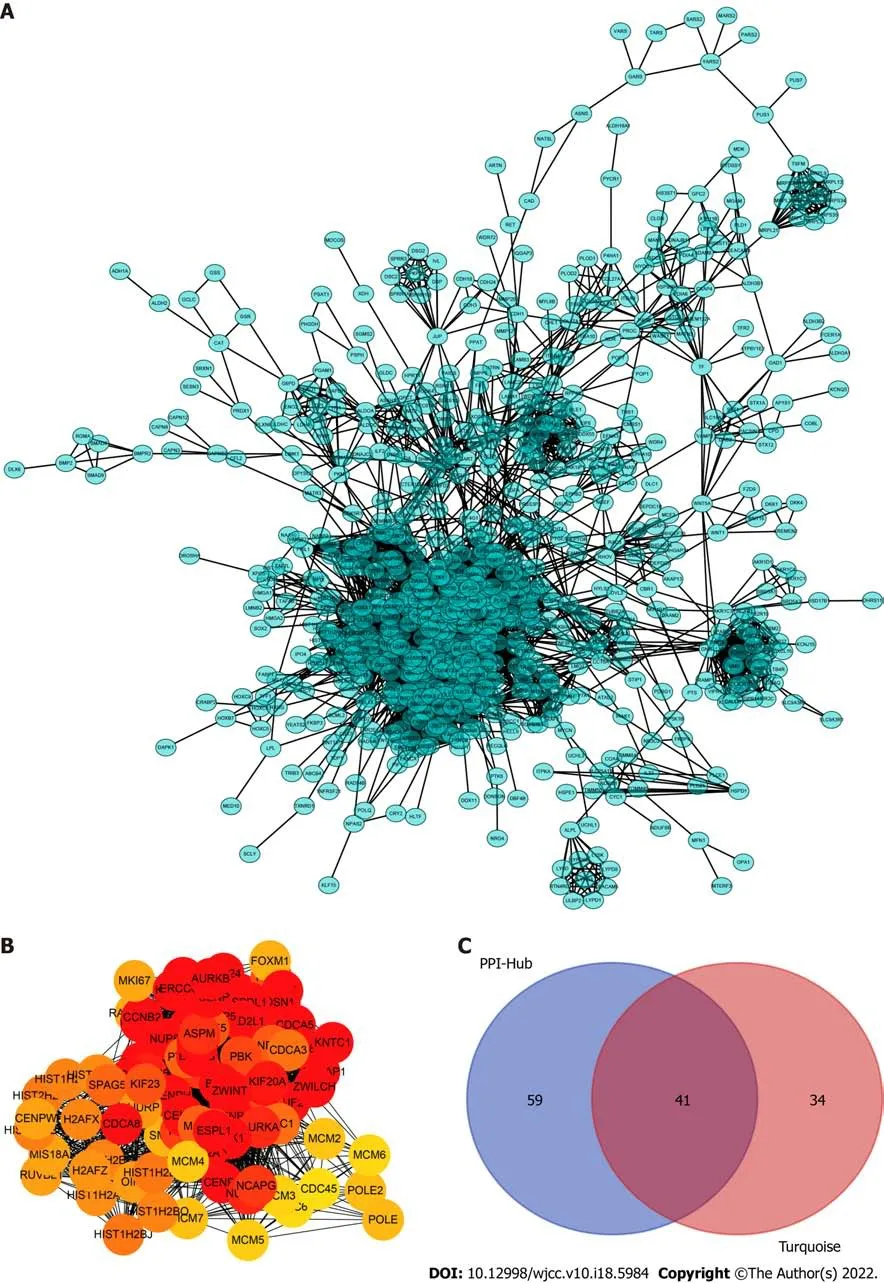
Protein–protein interaction network
We employed the STRING database to analyze the interaction between the module genes and set the confidence score to ≥ 0.9. The Cytohubba plug-in of Cytoscape software (version 3.7.0) was used to identify the core genes in the network.
The development of high-throughput technology has made important contributions to the identification of a large number of target genes in various diseases[7]. At the same time, as an emerging crossdiscipline, bioinformatics analysis is widely used in the discovery of disease-related genes, new drug molecular targets, drug design, and functional analysis, which is helpful for the discovery of disease mechanisms[8]. Xie
[9] performed bioinformatics analysis to analyze tumorigenesis-related genes and their target miRNAs in colon cancer, which facilitated the exploration of the potential targets for diagnosis, prognosis and treatment of colon carcinoma. Using RNA-Seq and bioinformatics methods,several key genes including
,
and
were identified in esophageal squamous cell carcinoma[10]. Many genes associated with LC progression and invasion have been identified by a combination of bioinformatics analysis and high-throughput sequencing[11-13].
Construction of predictive model
All data in this study were analyzed using GraphPad Prism 5 and R software. The data were expressed as mean ± SD. A two-tailed
test was applied for quantitative real-time polymerase chain reaction (qRTPCR) analysis among different groups. The results were considered to be significant at
< 0.05.
Cell culture
A549 and H1299 LC cells, and human lung fibroblasts were purchased from the Cancer Cell Repository(Shanghai Cell Bank, Shanghai, China). The medium used for cell culture was 10% Dulbecco’s modified eagle’s medium (supplemented with fetal bovine serum and penicillin/streptomycin). The cells were cultured in an incubator under 5% CO
at 37 ℃.
Quantitative real-time polymerase chain reaction
We use 1 mL TRIzol (Invitrogen, Grand Island, NY, United States) and 200 μL chloroform to extract the total RNA from 2 × 10
cells in LC cells. Total RNA was reverse transcribed into cDNA (TaKaRa Bio,Shiga, Japan). The cDNA, primers, and the SYBR Green PCR Master Mix (TOYOBO, Osaka, Japan) were quantitatively detected by PCR. The primer sequences of all genes are depicted in Table 1. The gene expression level was evaluated by the 2
method. All experiments were repeated three times.
Statistical analyses
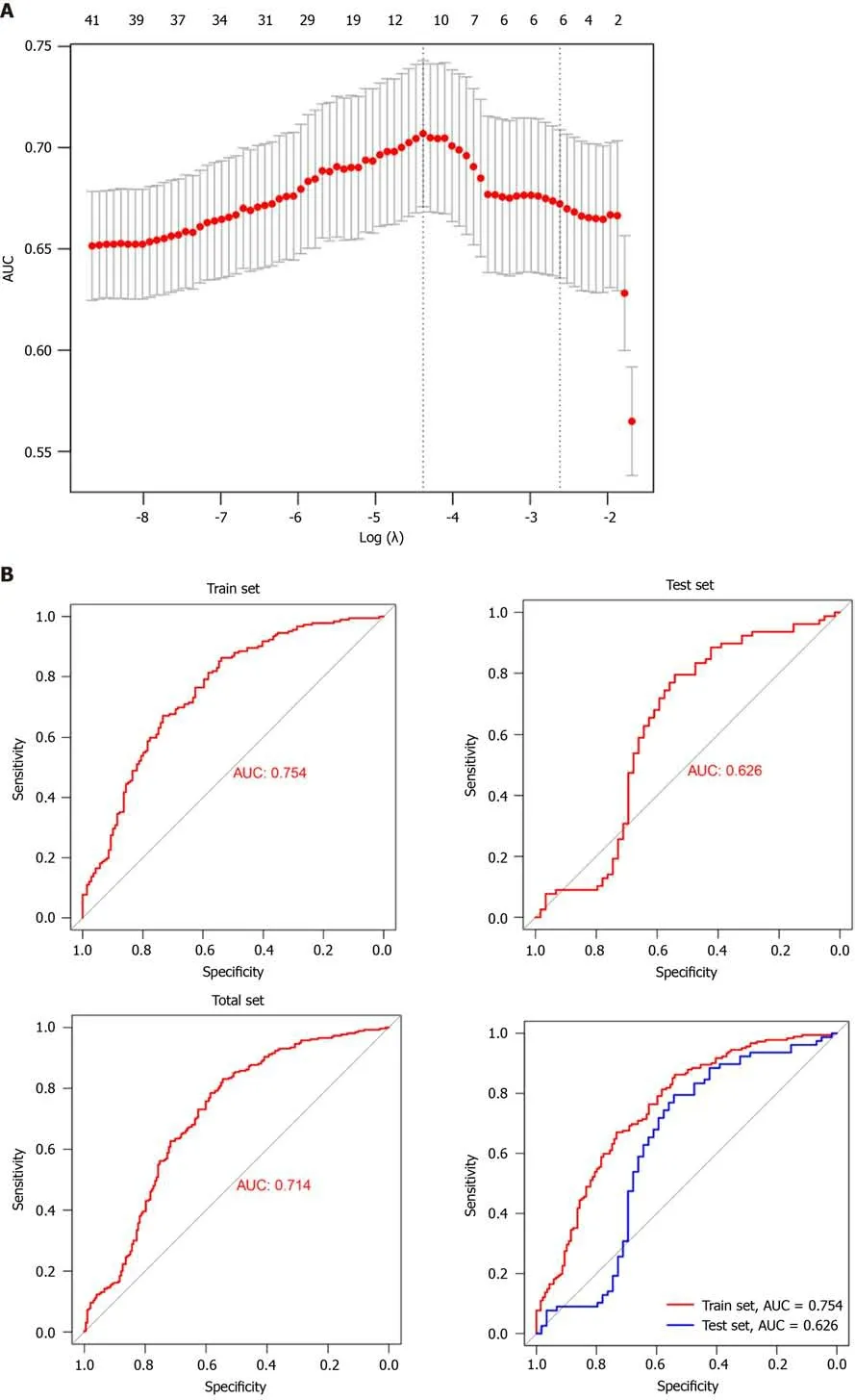
All patients were assigned to training and validation sets in the ratio of 6:4. The least absolute contraction and selection operator (LASSO) reduced the data dimensionality, and Cox regression analysis was applied to construct a patient prognostic evaluation model. The predictive efficacy of the model was evaluated by the receiver operating characteristic (ROC) curve. A nomogram was used to visualize the scoring system through the rms package in the R software.
RESULTS
Identification of intersecting genes between GEO cohort and TCGA dataset
We screened DEGs based on the TCGA dataset by including 1037
/108
samples. A total of 10970 DEGs were selected based on the criteria of
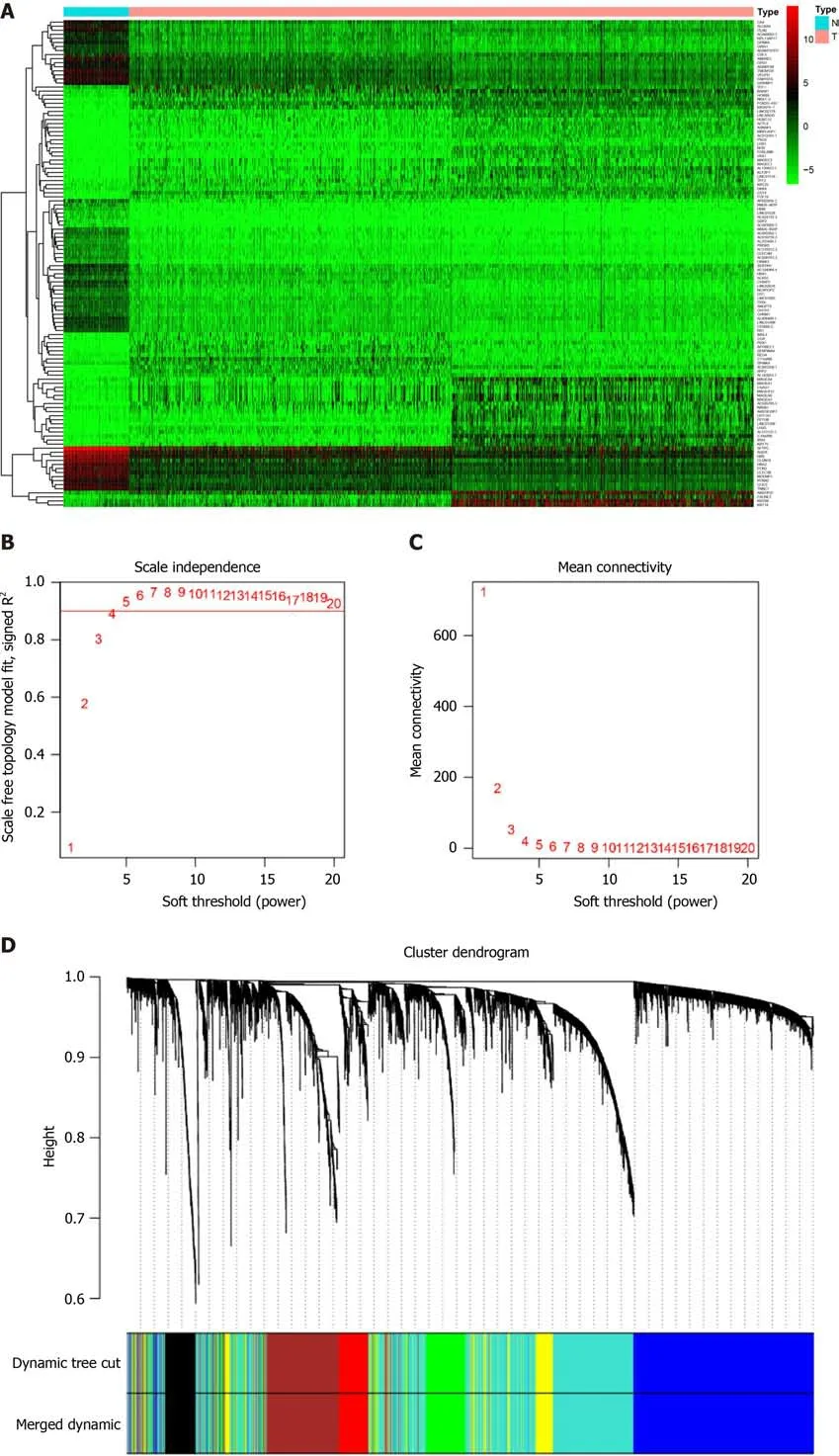
< 0.05 and |log 2 FC| > 1. The top 30 up- and downregulated genes are shown in Figure 1A. We then intersected DEGs of TCGA with all genes in the GEO dataset and found 5007 common genes for further WGCNA.

WGCNA
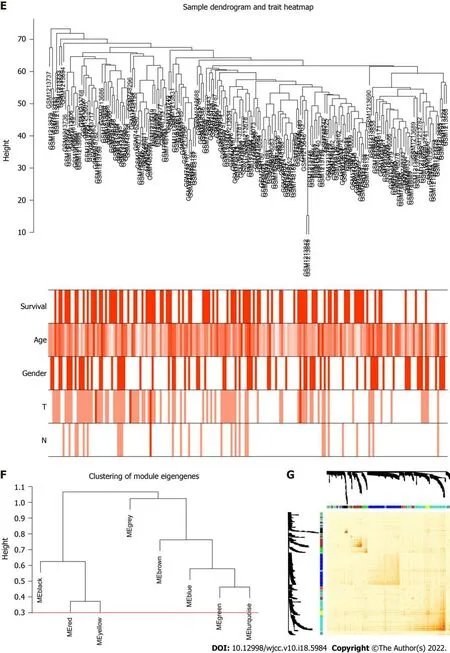
To determine the roles of common DEGs associated with prognosis and other clinicopathological characteristics of LC patients, WGCNA was performed to construct a coexpression network. As shown in Figure 1B and C, the correlation coefficient was converted to the adjacent coefficient according to the optimal parameter (β = 5). Thereafter, highly correlated samples and delete discrete samples were clustered (Figure 1E). A threshold of 0.25 and a minimum gene number of 150 were considered to merge similar modules. Figure 1D shows eight modules that were finally selected on the basis of the filter criteria. The hierarchical clustering of module hub genes is shown in Figure 1F.
Identification of highly correlated modules
本研究工具有四個。第一,自編調查問卷:分閉合式和開放式。閉合式題目采用五級量表形式,用因子分析、主成分分析法和Cronbach alpha系數檢驗問卷的信效度。以此測量學生的學習策略、學習需求、動機、情感策略、對課程的期望以及自我評價。第二,口語測試卷:每學期期末口語考試題和CET-SET4,用來測評學生的口語成績和水平。第三,聽力測試卷:每學期期末試卷和CET4聽力試卷,用來測評學生的聽力成績和水平。第四,聽說實踐作業展示:內容主要為時政焦點訪談、文化展示、商務會談等切合實際交際場景的主題,用來測評學生的語言綜合應用能力,尤其是口語表達能力。
GO and KEGG analyses in modules
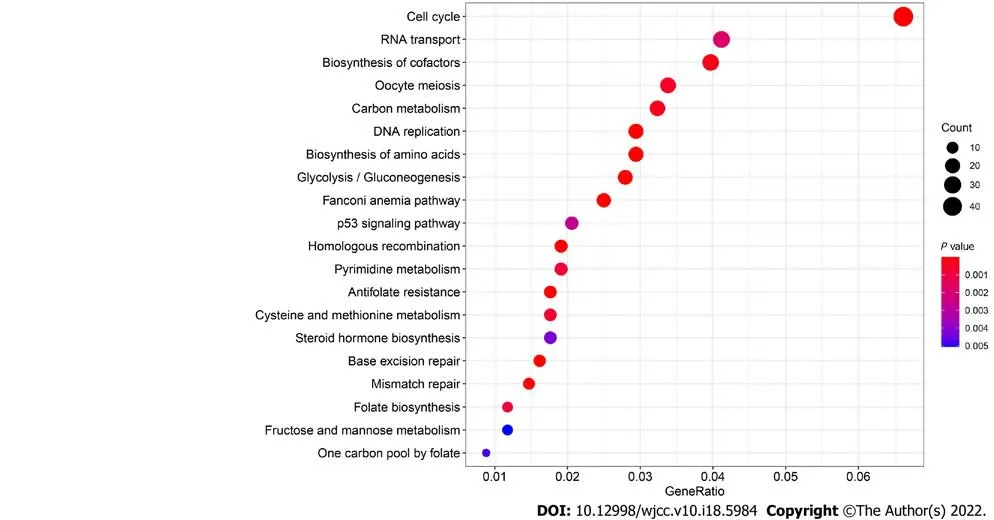
GO enrichment analysis experiments showed that the turquoise module genes mainly encoded for ATPase, helicase, 3′-5′ DNA helicase, DNA-dependent ATPase, DNA helicase, ATP-dependent DNA helicase, and ATP-dependent helicase and associated with the binding of many molecules including DNA replication origin, single-stranded DNA, and tubulin. KEGG analysis indicated that many signaling pathways involved in Fanconi anemia and the p53 signaling pathway were correlated with turquoise module genes. In addition, other important pathways such as metabolism of carbon,pyrimidine, cysteine, and methionine, cell cycle, and DNA repair were also found in the turquoise module. These results indicated that the mechanism that affects the survival of LC patients may be closely related to the molecular binding mechanism and several important signaling pathways(Figure 2D).
在給朱榮生、王子剛的復信中,劉少奇分別對兩人來信反映的問題進行了詳盡分析,著重批評了他們工作打不開局面的主觀原因(如朱榮生的顧此失彼、抓小棄大;王子剛強調兆征縣工作沒有基礎,卻不開展積極的思想斗爭以克服消極情緒,只在干部中打圈子),以及錯誤想法(如王子剛試圖用“稍微強迫一下子”的辦法要群眾當紅軍)。信中指出:只有改造了那個不健全的組織,在思想斗爭中團結與提拔了積極的干部,把組織健全起來,才能保證我們動員到廣大群眾中去,獲得很大的成績。
Establishment of protein–protein interaction networks and selection of module genes
由于煤燃燒產生的污染物主要是二氧化硫和一氧化碳,所以在此選擇二氧化硫為計算對象,煤炭中硫可分為兩部分,一部分叫做可燃性硫,它燃燒之后會釋放出二氧化硫,另一部分是不可燃性硫,顧名思義不可以燃燒直接歸為灰塵。根據相關化學方程式,可以得到以下關系:
Construction of the hub-genes-based scoring system
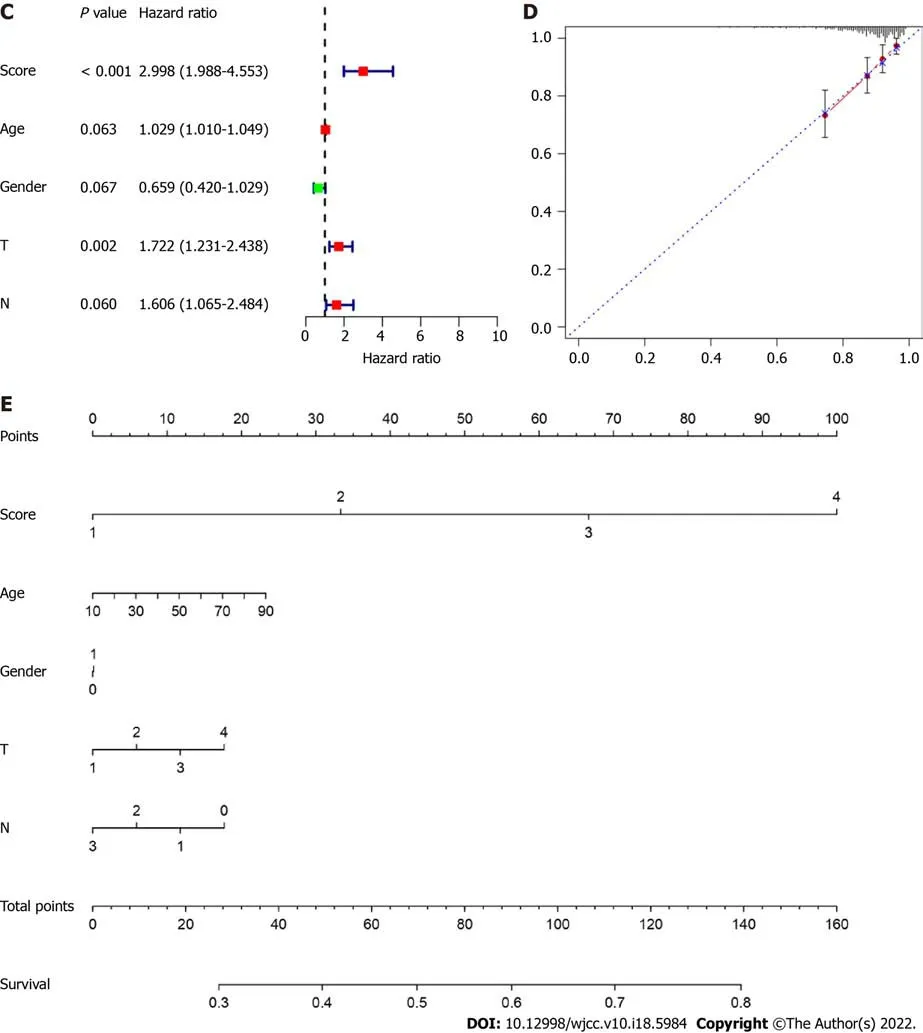
Subsequently, we performed a LASSO-logistic analysis of real hub genes to establish a prognostic evaluation model. Finally, 11 prognostic genes were selected in the predictive model in the training dataset, namely
,
,
,
,
,
,
,
,
,
,
and
(Figure 4A). The risk score = 4.43 (Intercept) + CCNB2-expression × 0.552 + CDC20-expression ×0.037 CENPO-expression × 0.287 + FOXM1-expression × 0.106 + HJURP-expression × 0.229 + NEK2-expression × 0.083 OIP5-expression × 0.020 PLK1-expression × 0.520 + PRC1-expression × 0.192 SKA1-expression × 0.110 + UBE2C-expression × 0.263. Subsequently, we evaluated the reliability of the model by the ROC curve. The results showed that the area under the curve of the training set and test set were 0.754 and 0.626, respectively (Figure 4B). Cox regression analysis showed that risk score was an independent risk factor for predicting the poor prognosis of LC patients (Figure 4C). To further evaluate the prognosis of LC, we constructed a nomogram based on risk factors (Figure 4D and E).
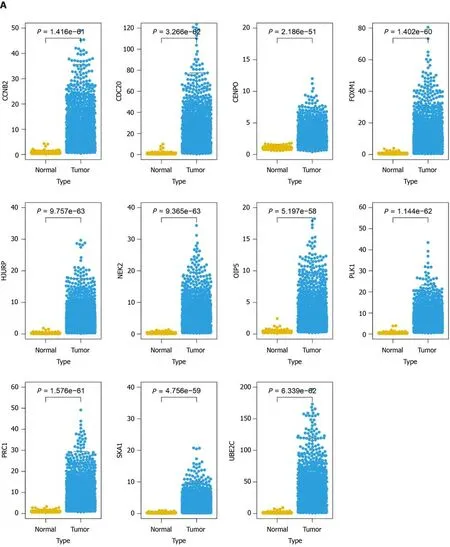
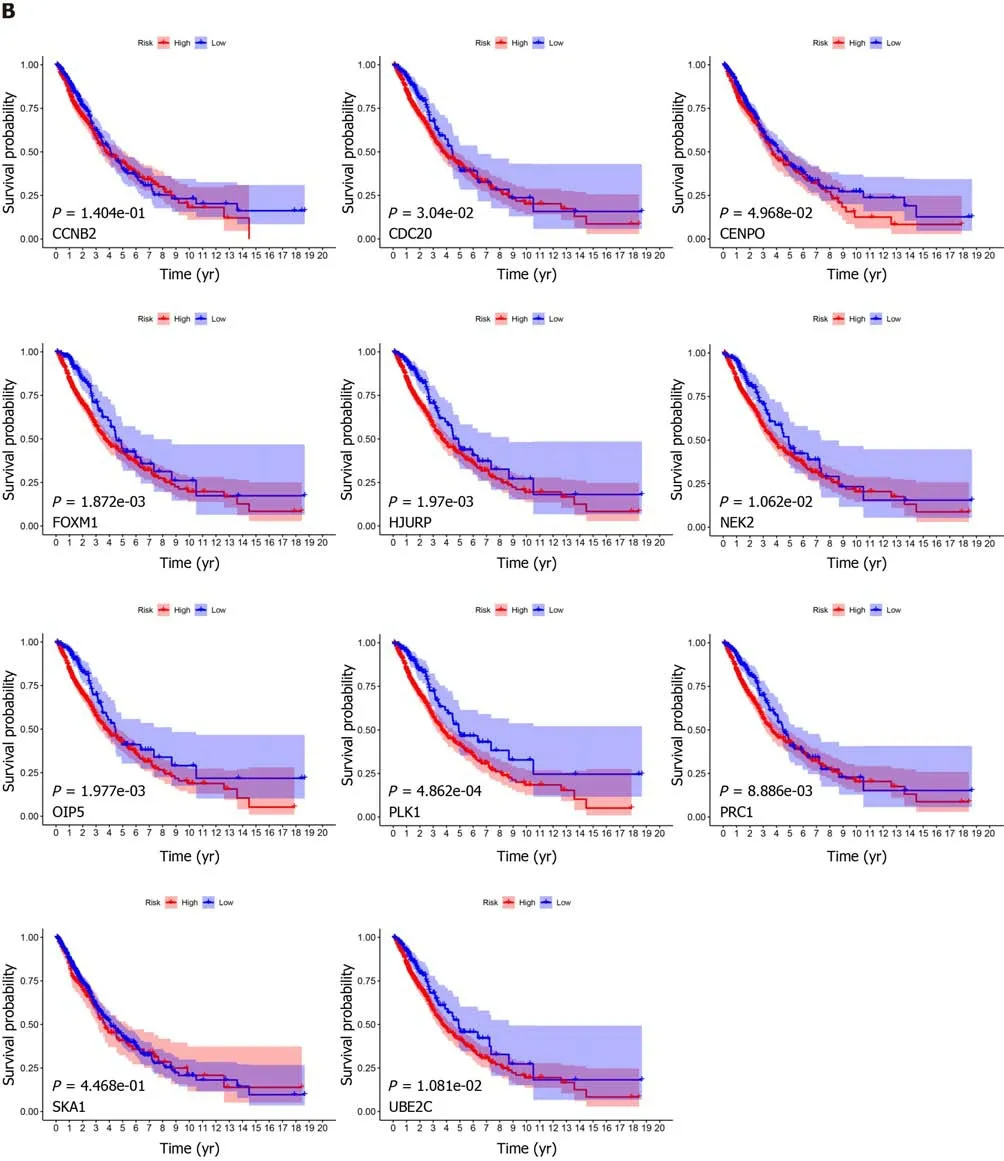
After the construction of the scoring system in the GEO dataset, we determined the effect of 11 genes in the TCGA dataset. As shown in Figure 5A, all genes were differentially expressed in LC patients compared with normal samples. Moreover, all genes were significantly correlated with patient prognosis except for
and
(Figure 5B).
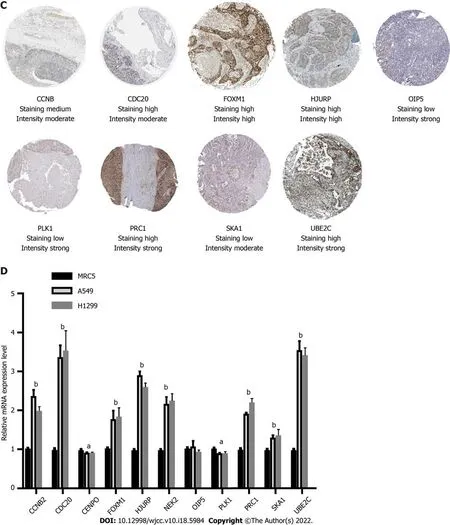
To validate the expression of the 11 hub genes, we performed an immunohistochemistry experiment obtained from the Protein Atlas database. Immunohistochemistry indicated that the protein levels of CDC20, FOXM1, HJURP, PRC1, UBE2C and CCNB2 were increased in the LC samples compared with the normal samples, whereas those of OIP5, PLK1 and SKA1 were decreased (Figure 5C). To explore the mRNA expression levels of these genes, we performed qRT-PCR analysis and found that the mRNA levels of
,
,
,
,
,
,
and
were increased in the LC cell lines, whereas those of
,
and
was decreased (Figure 5D).
To determine which cluster of genes in the turquoise module have a pivotal effect on the prognosis of LC, we constructed protein-protein interaction networks using the STRING database (Figure 3A) and Cytoscape software and found 100 hub genes located in the core area of the network (Figure 3B). We then intersected the 100 hub genes with 75 key genes sorted by MM to identify real hub genes associated with prognosis (Figure 3C).
DlSCUSSlON
LC treatments include a combination of radical surgery, radiation therapy, chemotherapy, and precise targeted therapy[14]. Despite advancement in LC treatment and diagnosis, the 5-year overall survival rate remains low[15]. The main reason is that patients who are diagnosed with advanced and metastasis LC cannot undergo radical surgery. Therefore, more specific and sensitive biomarkers are needed to facilitate early diagnosis and prediction of overall survival.
Recently, several therapeutic targets and prognostic biomarkers have been identified using advanced high-throughput sequencing technology and integrative bioinformatics analysis. Previous studies have reported many prognostic biomarkers in LC by performing a combined analysis using TCGA and GEO datasets and validated by
experiments. Sun
[16] reported the role of C-type lectin domain family 3 member B (CLEC3B) in tumor progression, prognosis, and immune responses in LC by performing RNA-Seq and bioinformatics analysis; the expression and methylation of CLEC3B were also validated by qRT-PCR analysis. miRNA-144-3p, an important noncoding RNA, was identified and validated as an independent risk factor for LC prognosis by performing bioinformatics analysis and qRT-PCR[17]. However, because of the insufficient sample size, biological heterogeneity, and different statistical methods, highly effective genes are not found in clinical practice. Moreover, the prediction efficiency in tumor patients could be limited by simply using a single GS instead of a multi-GS.Therefore, more biological markers and more effective prediction models are required for the prevention and treatment of LC.
In the present study, we initially detected DEGs based on the TCGA dataset and intersected these DEGs with all GEO cohort genes to obtain an expression profile. We used the WGCNA algorithm to identify core genes in GEO expression data and that were highly related to clinical features. WGCNA classified eight modules and subsequently correlated the modules with clinical characteristics. In these modules, the turquoise module contained 1673 genes that showed the highest correlation with LC patient prognosis. We then performed GO enrichment and KEGG pathway analyses of the turquoise module genes and found that the function of these genes was mainly related to activation of enzymes including ATPase, helicase, 3′-5′ DNA helicase, DNA-dependent ATPase, DNA helicase, ATPdependent DNA helicase, and ATP-dependent helicase and binding of many molecules including DNA replication origin, single-stranded DNA, and tubulin and activation of signaling pathways involved in Fanconi anemia and p53 signaling pathway. Subsequently, we performed a protein-protein interaction network analysis of the genes contained in the yellow module and intersected the network hub genes with MM > 0.8. Forty-one genes were selected and subjected to LASSO-logistic regression. We finally identified 11 prognostic genes, namely
,
,
,
,
,
,
,
,
,
,
and
. Among these genes,
and
are the most studied genes in LC. FOXM1, an important family member of the FOX family, plays a pivotal role in a series of biological processes, including facilitating cell proliferation, differentiation, and organ development[18]. FOXM1 level is significantly increased in LC cells and could be regulated by miR-216b, which promotes cancer progression and epithelial-mesenchymal transition in LC cells[19]. Moreover, FOXM1 could directly regulate the radiosensitivity of LC cells
interacting with KIF20A, suggesting that FOXM1 might be a novel therapeutic target for LC treatment. FOXM1 could also be regulated by other important molecules. The family with sequence similarity 188-member B is a member of the novel putative deubiquitinase family and directly binds to FOXM1, which promotes LC progression[20]. PLK1 is highly correlated with LC progression. PLK1 can target and regulate the transforming growth factor β signaling pathway, and then amplify its metastatic activity by positive feedback[20]. PLK1 is also regulated by long noncoding RNAs. For instance, miR-296-5p decreases the ability of cell invasion and migration by directly targeting PLK1 in LC cells[21]. However, other genes have not been actively researched, especially with regard to the mechanism of progression in LC. Therefore, in-depth knowledge of these genes will help develop new biomarkers for early LC diagnosis and prediction of prognosis.
After the scoring system was constructed, we further evaluated the performance of the model in LC patients. The ROC curve showed that the model had an excellent predictive performance. In addition,the risk predictor of the model can be considered an independent risk factor for predicting LC prognosis. To conclude, this study showed the potential of prognostic genes in LC patients using WGCNA combined with the established predictive model. However, this study also had some limitations. Firstly, LC patients were from public databases, thus the number of samples was limited. In future studies, we will collect samples from our hospital to expand the sample size to validate the predictive model. Second, the molecular biological mechanism by which the hub gene affects the prognosis of patient needs to be further explored.
CONCLUSlON
This study used the WGCNA algorithm to identify functional modules highly correlated with LC prognosis. After construction of the predictive model, we screened and validated 11 prognostic genes,which might be considered new therapeutic targets for the diagnosis and treatment of LC. This study also had some limitations. The mechanisms of the effect of the 11 prognostic genes on cancer progression need to be studied in the future.
ARTlCLE HlGHLlGHTS

FOOTNOTES
The authors have no conflicts of interest to declare.
This study was approved by the Ethics Committee of the Fenghua District People’s Hospital.
This study does not involve the clinical trials, so the clinical trial registration is not required.
The data that support the findings of current study are publicly available, so the signed informed consent document is not required.
Zhong C and Liang Y conceptualized and designed the article; Zhong C and Wang Q analyzed and interpreted the data; Zhong C drafted of the article; Liang Y and Tang HW were responsible for critical revision of the article for important intellectual content.
由圖11可知,模糊PID調平控制系統在2 s以內就能達到穩定,而且沒有超調,而常規PID控制在將近4 s時才能達到穩定,且超調量將近40%,仿真結果表明模糊PID控策略在混合臂高空作業車工作斗調平控制系統性能上優于常規PID控制策略,該模糊PID調平控制系統滿足要求,可用于實際需求。
No additional data are available.
然后進入任務實施階段。首先老師在線下針對學生在導學中提出的問題進行解答,然后發布任務操作視頻,學生在完成任務的過程中,可以反復查看,不斷完善任務。同時,要求學生提交相關任務完稿。為了更好的便于學生自主學習,在此過程中會開放相應的疑難討論論壇,供學生隨時提問討論。
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
China
Cheng Zhong 0000-0002-4077-2676; Yun Liang 0000-0003-3759-2997; Qun Wang 0000-0003-2258-4791;Hao-Wei Tan 0000-0001-6188-9294; Yan Liang 0000-0002-8608-0229.
紅琴與她那個見風就長的女兒在一起放風箏,與風影相比,她更是世俗的,紅塵世界里的平凡瑣事,也沒有走進傳奇故事里的必要與可能,更沒有佛門子弟的那種清苦與執著。紅琴是善于忘記的,很容易滿足的,風影則不同,他烙印在心底里的傷與痛是永遠不可能抹去的,而紅琴似乎已經淡忘了。風影站在草地上發呆,從眼神到心頭,都有一種說不清道不明的憂傷。
“你說你沒事結什么婚呵?”何西指著照片“我們科的丁主任,看上我了,非把他女兒介紹給我,還是跟我爸說的,我爸覺得我也該有女朋友了,就把我給安排了,明兒就得去見面,接頭暗號都定了。你說他長這樣,他女兒……”
Gong ZM
Kerr C
Gong ZM
1 Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012 . CA Cancer J Clin 2015 ;65 : 87 -108 [PMID: 25651787 DOI: 10 .3322 /caac.21262 ]
2 Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015 .
2016 ; 66 : 115 -132 [PMID: 26808342 DOI: 10 .3322 /caac.21338 ]
3 Villalobos P, Wistuba II. Lung Cancer Biomarkers. Hematol Oncol Clin North Am 2017 ; 31 : 13 -29 [PMID: 27912828 DOI: 10 .1016 /j.hoc.2016 .08 .006 ]
4 Di X, Jin X, Li R, Zhao M, Wang K. CircRNAs and lung cancer: Biomarkers and master regulators. Life Sci 2019 ; 220 :177 -185 [PMID: 30711537 DOI: 10 .1016 /j.lfs.2019 .01 .055 ]
5 Ciliberto D, Staropoli N, Caglioti F, Gualtieri S, Fiorillo L, Chiellino S, De Angelis AM, Mendicino F, Botta C, Caraglia M, Tassone P, Tagliaferri P. A systematic review and meta-analysis of randomized trials on the role of targeted therapy in the management of advanced gastric cancer: Evidence does not translate?
2015 ; 16 : 1148 -1159 [PMID:26061272 DOI: 10 .1080 /15384047 .2015 .1056415 ]
6 Tsoukalas N, Kiakou M, Tsapakidis K, Tolia M, Aravantinou-Fatorou E, Baxevanos P, Kyrgias G, Theocharis S. PD-1 and PD-L1 as immunotherapy targets and biomarkers in non-small cell lung cancer. J BUON 2019 ; 24 : 883 -888 [PMID:31424637 ]
7 Moncho-Amor V, Pintado-Berninches L, Iba?ez de Cáceres I, Martín-Villar E, Quintanilla M, Chakravarty P, Cortes-Sempere M, Fernández-Varas B, Rodriguez-Antolín C, de Castro J, Sastre L, Perona R. Role of Dusp6 Phosphatase as a Tumor Suppressor in Non-Small Cell Lung Cancer.
2019 ; 20 [PMID: 31027181 DOI: 10 .3390 /ijms20082036 ]
8 Kulasingam V, Diamandis EP. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies.
2008 ; 5 : 588 -599 [PMID: 18695711 DOI: 10 .1038 /ncponc1187 ]
9 Xie B, Zhao R, Bai B, Wu Y, Xu Y, Lu S, Fang Y, Wang Z, Maswikiti EP, Zhou X, Pan H, Han W. Identification of key tumorigenesisrelated genes and their microRNAs in colon cancer.
2018 ; 40 : 3551 -3560 [PMID: 30272358 DOI:10 .3892 /or.2018 .6726 ]
10 Zhao Y, Zhu J, Shi B, Wang X, Lu Q, Li C, Chen H. The transcription factor LEF1 promotes tumorigenicity and activates the TGF-β signaling pathway in esophageal squamous cell carcinoma.
2019 ; 38 : 304 [PMID:31296250 DOI: 10 .1186 /s13046 -019 -1296 -7 ]
11 Zhao T, Khadka VS, Deng Y. Identification of lncRNA biomarkers for lung cancer through integrative cross-platform data analyses.
2020 ; 12 : 14506 -14527 [PMID: 32675385 DOI: 10 .18632 /aging.103496 ]
12 Xue L, Xie L, Song X. Identification of potential tumor-educated platelets RNA biomarkers in non-small-cell lung cancer by integrated bioinformatical analysis.
2018 ; 32 : e22450 [PMID: 29665143 DOI: 10 .1002 /jcla.22450 ]
13 Liu L, Ahmed T, Petty WJ, Grant S, Ruiz J, Lycan TW, Topaloglu U, Chou PC, Miller LD, Hawkins GA, Alexander-Miller MA, O'Neill SS, Powell BL, D'Agostino RB Jr, Munden RF, Pasche B, Zhang W. SMARCA4 mutations in KRASmutant lung adenocarcinoma: a multi-cohort analysis.
2021 ; 15 : 462 -472 [PMID: 33107184 DOI:10 .1002 /1878 -0261 .12831 ]
14 Hensing T, Chawla A, Batra R, Salgia R. A personalized treatment for lung cancer: molecular pathways, targeted therapies,and genomic characterization.
2014 ; 799 : 85 -117 [PMID: 24292963 DOI:10 .1007 /978 -1 -4614 -8778 -4 _5 ]
15 Li F, He H, Qiu B, Ji Y, Sun K, Xue Q, Guo W, Wang D, Zhao J, Mao Y, Mu J, Gao S. Clinicopathological characteristics and prognosis of lung cancer in young patients aged 30 years and younger. J Thorac Dis 2019 ; 11 : 4282 -4291 [PMID:31737313 DOI: 10 .21037 /jtd.2019 .09 .60 ]
16 Sun J, Xie T, Jamal M, Tu Z, Li X, Wu Y, Li J, Zhang Q, Huang X. CLEC3 B as a potential diagnostic and prognostic biomarker in lung cancer and association with the immune microenvironment.
2020 ; 20 : 106 [PMID:32265595 DOI: 10 .1186 /s12935 -020 -01183 -1 ]
17 Chen YJ, Guo YN, Shi K, Huang HM, Huang SP, Xu WQ, Li ZY, Wei KL, Gan TQ, Chen G. Down-regulation of microRNA-144 -3 p and its clinical value in non-small cell lung cancer: a comprehensive analysis based on microarray,miRNA-sequencing, and quantitative real-time PCR data.
2019 ; 20 : 48 [PMID: 30832674 DOI:10 .1186 /s12931 -019 -0994 -1 ]
18 Zhang Y, Qiao WB, Shan L. Expression and functional characterization of FOXM1 in non-small cell lung cancer.
2018 ; 11 : 3385 -3393 [PMID: 29928129 DOI: 10 .2147 /OTT.S162523 ]
19 Wang L, Wang Y, Du X, Yao Y, Wang L, Jia Y. MiR-216 b suppresses cell proliferation, migration, invasion, and epithelial-mesenchymal transition by regulating FOXM1 expression in human non-small cell lung cancer.
2019 ; 12 : 2999 -3009 [PMID: 31114243 DOI: 10 .2147 /OTT.S202523 ]
20 Choi YE, Madhi H, Kim H, Lee JS, Kim MH, Kim YN, Goh SH. FAM188 B Expression Is Critical for Cell Growth
FOXM1 Regulation in Lung Cancer. Biomedicines 2020 ; 8 [PMID: 33142744 DOI: 10 .3390 /biomedicines8110465 ]
21 Xu C, Li S, Chen T, Hu H, Ding C, Xu Z, Chen J, Liu Z, Lei Z, Zhang HT, Li C, Zhao J. miR-296 -5 p suppresses cell viability by directly targeting PLK1 in non-small cell lung cancer. Oncol Rep 2016 ; 35 : 497 -503 [PMID: 26549165 DOI:10 .3892 /or.2015 .4392 ]
猜你喜歡
作文大王·笑話大王(2021年4期)2021-04-26 19:00:35
英語文摘(2020年9期)2020-11-26 08:10:12
甘肅教育(2020年6期)2020-09-11 07:45:16
甘肅教育(2020年22期)2020-04-13 08:10:54
甘肅教育(2020年20期)2020-04-13 08:04:42
當代陜西(2019年5期)2019-11-17 04:27:32
電影(2018年9期)2018-11-14 06:57:21
作文世界(小學版)(2018年4期)2018-10-16 17:13:34
快樂作文·低年級(2016年12期)2017-01-03 20:52:44
快樂作文·低年級(2016年6期)2016-06-24 18:58:40
 World Journal of Clinical Cases2022年18期
World Journal of Clinical Cases2022年18期
- World Journal of Clinical Cases的其它文章
- Stem cells as an option for the treatment of COVID-19
- Development of clustered regularly interspaced short palindromic repeats/CRISPR-associated technology for potential clinical applications
- Prostate sclerosing adenopathy: A clinicopathological and immunohistochemical study of twelve patients
- Effectiveness and postoperative rehabilitation of one-stage combined anterior-posterior surgery for severe thoracolumbar fractures with spinal cord injury
- Identification of potential key molecules and signaling pathways for psoriasis based on weighted gene coexpression network analysis
- Coinfection of Streptococcus suis and Nocardia asiatica in the human central nervous system: A case report