有機污染物的高鐵酸鹽氧化去除強化技術研究進展
2022-06-22 08:32:06程和發(fā)
環(huán)境科學研究 2022年6期
李 宇,程和發(fā)
北京大學城市與環(huán)境學院,地表過程分析與模擬教育部重點實驗室,北京 100871
對于水中有機污染物的常用處理方法包括物理吸附法、化學氧化法和生物降解法,其中使用高鐵酸鹽〔Fe(Ⅵ)〕進行化學氧化是去除有機污染物的有效方法之一. Fe(Ⅵ)具有強氧化性,在酸性條件下的氧化還原電位(2.2 V)高于臭氧(O)、過氧化氫(HO)、氯氣(Cl)、二氧化氯(ClO)和高錳酸鉀(KMnO)等常用水處理氧化劑,并且在較寬的pH 范圍內具有較強的氧化能力. 相比于基于無選擇性的羥基自由基(OH)的高級氧化技術(AOPs),Fe(Ⅵ)的選擇性高,易于氧化富含供電子基團的有機污染物,受環(huán)境基質影響較小,Fe(Ⅵ)與Cl、HCO、NO、SO等常見陰離子和Na、K等常見陽離子的反應活性均較低,低濃度的腐殖酸(HA)還能對Fe(Ⅵ)氧化有機污染物起到促進作用,且Fe(Ⅵ)能夠在實際污水處理的pH (pH 為6.0~9.0)下進行反應. 雖然Fe(Ⅵ)能夠有效氧化分子結構中含有不飽和官能團(如酚羥基、芳胺基和碳碳雙鍵)的有機污染物,但其在酸性條件下易于自分解,且選擇性高,因此Fe(Ⅵ)對許多不飽和度較低的有機污染物反應活性低. 此外,在堿性條件下,Fe(Ⅵ)氧化能力也會降低. 因此,Fe(Ⅵ)直接氧化法亟需改進,以擴大其適用范圍、提高其氧化能力,使其適用于實際污水處理.
近年來,國內外學者開發(fā)了諸多Fe(Ⅵ)氧化的強化技術,以提高Fe(Ⅵ)對有機污染物的處理效能,去除Fe(Ⅵ)本身難以氧化的有機污染物,并取得了較大進展. 根據Fe(Ⅵ)氧化的強化體系所產生的活性物種的類別,可將其分為基于高價鐵氧中間體〔Fe(Ⅳ)/Fe(Ⅴ)〕的Fe(Ⅵ)活化技術和基于活性自由基(如OH 和SO)等其他活性物種的Fe(Ⅵ)協(xié)同技術. Fe(Ⅵ)的活化及協(xié)同技術可以提高Fe(Ⅵ)與有機污染物的反應速率、Fe(Ⅵ)的利用效率和有機污染物的降解率,有效去除與Fe(Ⅵ)反應活性低的有機污染物,同時減少Fe(Ⅵ)用量. 但由于在Fe(Ⅵ)活化或協(xié)同體系中起作用的活性物種發(fā)生變化,進而改變有機污染物的降解動力學和降解機理,以及降解產物的種類、分布和毒性,因此需要全面深入地對Fe(Ⅵ)的活化及協(xié)同技術進行評估和研究. 在已有研究基礎上,該文對近年來開發(fā)的Fe(Ⅵ)活化及協(xié)同技術的研究進展進行系統(tǒng)性總結,梳理Fe(Ⅵ)直接氧化有機污染物的處理效能、反應動力學和氧化機理、Fe(Ⅵ)的活化技術及協(xié)同技術氧化有機污染物的效能與機理,討論Fe(Ⅵ)活化及協(xié)同技術引起的有機污染物降解產物種類、分布及毒性變化,并對未來研究方向進行展望,以期為Fe(Ⅵ)在實際水處理中的高效利用提供參考方法.
1 Fe(Ⅵ)直接氧化有機污染物
1.1 處理效能
Fe(Ⅵ)直接氧化法已經廣泛用于去除水中有機污染物,并展現出良好的處理效能. Sun 等在pH=8.0 時使用30 μmol/L 的Fe(Ⅵ)氧化降解2 μmol/L的苯胺,發(fā)現150 s 時即可達到92%的去除率;楊濱等在pH=7.0 時使用Fe(Ⅵ)氧化降解三氯生,發(fā)現Fe(Ⅵ)與三氯生的濃度比大于7∶1 時,2 μmol/L 的三氯生即可得到完全去除;黃軍磊等在pH=7.0 時使用300 μmol/L 的Fe(Ⅵ)氧化降解20 μmol/L 的吲哚美辛,發(fā)現20 min 時可達到95%的去除率;Feng等在pH=7.0 時使用Fe(Ⅵ)氧化降解30 μmol/L 的氟喹諾酮類抗生素,發(fā)現在Fe(Ⅵ)與目標污染物的濃度比為20∶1 時,在2 min 內即可完全去除恩諾沙星、諾氟沙星、氧氟沙星和馬波沙星4 種抗生素. 綜上,Fe(Ⅵ)可在數分鐘內基本完全去除含有供電子基團的有機污染物,具有良好的處理效能.
1.2 反應動力學
Fe(Ⅵ)和大部分有機污染物(以X 表示)的氧化還原反應都遵循二級反應動力學模型,即
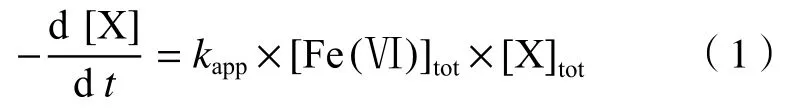
式中:為Fe(Ⅵ)與有機污染物反應的表觀二級動力學反應速率常數,L/(mol·s);[Fe(Ⅵ)]為不同離子形態(tài)Fe(Ⅵ)的總濃度,mol/L;[X]為不同離子形態(tài)有機污染物的總濃度,mol/L. Fe(Ⅵ)的解離常數(p)為1.6、3.5 和7.2,主要以HFeO、HFeO、HFeO和FeO四種形態(tài)存在.通常是在Fe(Ⅵ)濃度遠低于有機污染物濃度的條件下,使Fe(Ⅵ)濃度變化符合一級動力學特征,使用停流光譜技術測定Fe(Ⅵ)濃度的變化速率,進而計算得到的,一般在10~10L/(mol·s)范圍內.
Fe(Ⅵ)氧化取代基不同的苯環(huán)取代化合物時,與取代基之間存在結構-活性關系. 例如,Sun等研究表明,Fe(Ⅵ)和取代苯胺化合物反應的的對數值與取代基系數呈線性關系;Fe(Ⅵ)氧化苯并三唑類化合物和多氯聯(lián)苯硫化物的的對數值均與取代基系數存在線性自由能關系. 通過Fe(Ⅵ)和苯環(huán)取代化合物反應的的對數值與取代基系數之間的線性自由能關系,可以使用已知Fe(Ⅵ)與苯環(huán)取代化合物的和取代基系數等參數,預測Fe(Ⅵ)與其他結構類似但取代基不同的苯環(huán)取代化合物反應的.
1.3 氧化機理
Fe(Ⅵ)對有機污染物的直接氧化遵循單電子(1 e)或雙電子(2 e)轉移機制,因此其對有機污染物的氧化具有高選擇性,電子轉移機理如圖1 所示.Fe(Ⅵ)的選擇性氧化導致其反應活性受到目標有機污染物的結構與性質的影響,Fe(Ⅵ)易于和含有富電子官能團的有機物(如有機硫化合物、胺類化合物、酚類化合物等)發(fā)生反應,但對于醇類化合物、醛類化合物、糖類化合物等反應活性較低. Fe(Ⅵ)氧化有機污染物的選擇性可以體現在Fe(Ⅵ)與有機污染物反應的的差異上,如Xie 等對比研究了Fe(Ⅵ)氧化兩種苯胂酸類化合物對氨基苯胂酸和洛克沙胂的動力學特征,發(fā)現分子中含有供電子基團氨基的對氨基苯胂酸與Fe(Ⅵ)反應的遠高于分子中含有吸電子基團硝基的洛克沙胂. Jiang 等使用Fe(Ⅵ)處理環(huán)丙沙星和布洛芬的混合溶液,發(fā)現環(huán)丙沙星的去除率明顯高于布洛芬,這是由于環(huán)丙沙星分子中的供電子基團更多. Yang 等使用Fe(Ⅵ)處理污水處理廠二級出水中的68 種有機污染物,發(fā)現Fe(Ⅵ)能夠選擇性地氧化降解富含供電子基團的有機污染物,如具有酚類結構的雌激素和三氯生,具有雙鍵結構的卡馬西平、雄激素、孕激素和糖皮質激素,以及含仲銨基的酸性藥物和含苯胺基的抗生素,而Fe(Ⅵ)與布洛芬、非諾洛芬、環(huán)磷酰胺等供電子基團較少的有機污染物的反應活性較低.

圖 1 Fe(Ⅵ)直接氧化有機污染物(以X 表示)的電子轉移機理示意[16]Fig.1 Diagram of the electron transfer mechanism during the oxidation of organic pollutant (represented by X) by Fe(Ⅵ)[16]
與有機污染物反應時,Fe(Ⅵ)首先被還原為中間價態(tài)的Fe(Ⅳ)/Fe(Ⅴ),Fe(Ⅳ)/Fe(Ⅴ)性質不穩(wěn)定,會快速自分解或進一步通過電子轉移與有機污染物反應.Fe(Ⅵ)與有機污染物的反應機理通常包括:①先通過單電子轉移從Fe(Ⅵ)轉化為Fe(Ⅴ),之后Fe(Ⅴ)進一步氧化有機污染物;②通過雙電子轉移轉化為Fe(Ⅳ)和二聚體,之后Fe(Ⅳ)進一步氧化有機污染物;③通過氧原子轉移(OAT)生成Fe(Ⅳ)和加氧產物,之后Fe(Ⅳ)進一步氧化有機污染物. 此外,Fe(Ⅵ)、Fe(Ⅴ)、Fe(Ⅳ)也會發(fā)生自分解反應,最終生成Fe(Ⅱ)和Fe(Ⅲ). 有機污染物被Fe(Ⅵ)氧化的降解路徑取決于其具體化學結構,不同的化學結構和官能團導致其降解機理和降解路徑不同,需要通過O 示蹤法和產物分析法對Fe(Ⅵ)氧化有機污染物的機理進行深入研究.
Fe(Ⅵ)直接氧化法對含有不飽和官能團的有機污染物的氧化能力較強,但存在一定局限性,如在酸性條件下Fe(Ⅵ)會迅速自分解,而在堿性條件下Fe(Ⅵ)的高穩(wěn)定性導致有機污染物降解緩慢. 此外,Fe(Ⅵ)氧化的高選擇性導致其與不飽和官能團較少的有機污染物的反應活性較低. 因此,Fe(Ⅵ)直接氧化法亟需進行改進與強化,從而開發(fā)出一系列Fe(Ⅵ)的活化及協(xié)同技術.
2 Fe(Ⅵ)活化技術
研究表明,中間價態(tài)的Fe(Ⅳ)/Fe(Ⅴ)與有機化合物反應的比Fe(Ⅵ)高2~6 個數量級,因此可以使用化學方法將Fe(Ⅵ)轉化為Fe(Ⅳ)/Fe(Ⅴ)以促進有機污染物的降解. Fe(Ⅵ)的活化技術通過在中性或偏堿性條件下,向Fe(Ⅵ)氧化體系中投加活化劑,誘導低活性的Fe(Ⅵ)生成高活性的Fe(Ⅳ)/Fe(Ⅴ),從而提高有機污染物的降解速率和降解率. 根據投加的活化劑在反應體系中的狀態(tài),可將Fe(Ⅵ)活化技術分為均相活化技術和異相活化技術.
2.1 均相活化技術
Fe(Ⅵ)的均相活化技術包括酸活化Fe(Ⅵ)、還原劑活化Fe(Ⅵ)等.
2.1.1 酸活化Fe(Ⅵ)
Manoli 等研究發(fā)現,向偏堿性的Fe(Ⅵ)和有機污染物混合體系中加入少量酸時,pH 變化很小,但咖啡因、安賽蜜、阿替洛爾的降解率提高了30%左右,并且所需反應時間大幅降低,他們推測酸活化Fe(Ⅵ)降解有機污染物的活性物種為Fe(Ⅳ)/Fe(Ⅴ).Manoli 等還發(fā)現,Fe(Ⅵ)氧化降解咖啡因受到水中Ca和Mg的抑制作用,而酸活化Fe(Ⅵ)可以減輕Ca和Mg對咖啡因降解的抑制作用,雖然酸活化前后咖啡因的降解都受到天然有機物(NOM)和二級污水的抑制作用,但Fe(Ⅵ)直接氧化咖啡因并不礦化,而酸活化Fe(Ⅵ)在NOM 存在和二級污水反應體系中均有明顯礦化. Manoli 等使用酸活化Fe(Ⅵ)處理實際污水處理廠出水中的多種有機污染物,發(fā)現相比于Fe(Ⅵ)直接氧化,污水中藥物的去除率提高了12.6%~56.2%,因此酸活化Fe(Ⅵ)技術更適用于實際水處理. 然而,以上研究均未直接觀測到酸活化Fe(Ⅵ)后產生的活性物種,因此酸活化Fe(Ⅵ)生成Fe(Ⅳ)/Fe(Ⅴ)的假設還需進一步試驗驗證.
2.1.2 還原劑活化Fe(Ⅵ)
由于過渡金屬元素Fe 具有多種價態(tài)(0、+2、+3、+4、+5 和+6),因此可以通過還原劑的還原作用將Fe(Ⅵ)轉化為強氧化性的Fe(Ⅳ)/Fe(Ⅴ),促進有機污染物的降解. Feng 等研究了多種無機還原劑對Fe(Ⅵ)的活化作用,發(fā)現還原劑可以在30 s 內有效活化Fe(Ⅵ)降解有機污染物,且降解效能與還原劑的種類和用量密切相關. Fe(Ⅵ)直接氧化甲氧芐氨嘧啶的降解率為16%,雙電子轉移還原劑〔如NHOH、As(Ⅲ)、Se(Ⅳ)、P(Ⅲ)和NO〕可 將Fe(Ⅵ)還 原 為Fe(Ⅳ),將降解率提高至20%~40%,而單電子轉移還原劑(如SO)則將Fe(Ⅵ)還原為Fe(Ⅴ),將降解率提高到100%. Fe(Ⅱ)、Mn(Ⅱ)、Fe(Ⅲ)等還原劑也可以有效活化Fe(Ⅵ),實現對有機污染物的高效去除. Dong 等研究發(fā)現,2,2-聯(lián)氮-二(3-乙基-苯并噻唑-6-磺酸)二銨鹽(ABTS)可以在較寬的pH 范圍(6.0~10.0)內活化Fe(Ⅵ)生成Fe(Ⅴ)和氧化性的ABTS,促進雙氯芬酸的降解. pH=8.0 時,Fe(Ⅵ)直接氧化體系和Fe(Ⅵ)/ABTS 體系降解雙氯芬酸的準一級降解速率分別為0.085 和3.08 min. 在pH 為7.0~9.0 范圍內,HO也可以作為還原劑,通過雙電子轉移活化Fe(Ⅵ)生成Fe(Ⅳ)〔Fe(Ⅵ)+HO→Fe(Ⅳ)+O,=21.18 L/(mol·s) (pH=8.0)〕,并通過鏈式反應生成Fe(Ⅴ),促進多種有機污染物的降解,其機理如圖2所示. pH=8.0 時,Fe(Ⅵ)直接氧化體系和Fe(Ⅵ)/HO體系降解磺胺甲惡唑的準一級降解速率分別為0.08和0.30 min.

圖 2 H2O2 活化Fe(Ⅵ)機理示意[32]Fig.2 Diagram of the mechanisms of Fe(Ⅵ) activation by H2O2[32]
相比于Fe(Ⅵ)直接氧化,還原劑活化Fe(Ⅵ)大多只能在反應初始階段促進有機污染物的氧化降解,且還原劑自身也會消耗產生的Fe(Ⅳ)/Fe(Ⅴ),因此Fe(Ⅵ)的整體氧化容量可能反而降低. 與其他還原劑不同,氨氮可以提高Fe(Ⅵ)與有機污染物的反應速率和有機污染物降解的表觀速率,而不只是在初始階段提高有機污染物的降解率. Feng 等研究發(fā)現,向Fe(Ⅵ)氧化體系加入氨氮后,Fe(Ⅵ)氧化氟甲喹的速率可以提高5~12 倍,具體數值取決于氨氮的濃度和pH. 這可能是由于氨氮可以通過與Fe(Ⅵ)的中間產物Fe(Ⅳ)/Fe(Ⅴ)絡合,提高Fe(Ⅳ)/Fe(Ⅴ)的反應活性,從而提高氟甲喹的準一級降解速率.
2.2 異相活化技術
異相活化技術是指向Fe(Ⅵ)與目標有機污染物的混合體系中投加不溶于水的固體顆粒,通過一系列固體表面作用或緩釋活化劑實現活化Fe(Ⅵ)的技術,已開發(fā)的異相活化技術包括二氧化硅(SiO)活化Fe(Ⅵ)、過氧化鈣(CaO)活化Fe(Ⅵ)、碳質材料活化Fe(Ⅵ)等.
2.2.1 SiO活化Fe(Ⅵ)
Manoli 等研究發(fā)現,在pH 約為8.0 時,使用4 g/L 的SiO可將Fe(Ⅵ)直接氧化咖啡因的降解率由53.0%升至100%,且增加SiO用量可以減輕NOM對咖啡因降解的抑制作用. 活化機理可能為SiO抑制Fe(Ⅱ)/Fe(Ⅲ)和Fe(Ⅵ)/Fe(Ⅴ)的反應,從而增加Fe(Ⅵ)的暴露量,使更多的Fe(Ⅵ)能與有機污染物反應. 此外,Fe(Ⅵ)可能會吸附在SiO凝膠表面,Fe 與Si 的作用能夠產生強氧化劑〔如Fe(Ⅳ)〕,并且降低Fe(Ⅵ)的自分解,從而促進咖啡因的氧化.
2.2.2 CaO活化Fe(Ⅵ)
Zhang 等使用CaO緩釋HO,以活化Fe(Ⅵ)降解磺胺甲惡唑,發(fā)現磺胺甲惡唑在Fe(Ⅵ)直接氧化體系和Fe(Ⅵ)/CaO體系中的降解率分別為35.4%和82.7%. 促進效果可能是由于CaO緩釋HO,一方面將Fe(Ⅵ)活化為Fe(Ⅳ),并通過鏈式反應生成Fe(Ⅴ);另一方面抑制了HO與Fe(Ⅳ)/Fe(Ⅴ)的副反應〔Fe(Ⅳ)+HO→Fe(Ⅱ)+O,=10L/(mol·s) (pH=7.0);Fe(Ⅴ)+HO→Fe(Ⅲ)+O,=5.07×10L/(mol·s) (pH=8.0)〕,從而提高了Fe(Ⅵ)和HO的利用率,促進有機污染物的降解. 此外,Fe(Ⅳ)/Fe(Ⅴ)在偏堿性條件下的自分解符合二級動力學特征,即Fe(Ⅳ)/Fe(Ⅴ)的濃度越高,其自分解越劇烈,因此CaO可以通過緩釋HO活化Fe(Ⅵ),降低Fe(Ⅳ)/Fe(Ⅴ)的濃度,從而抑制其自分解,提高有機污染物的降解率.
2.2.3 碳質材料活化Fe(Ⅵ)
碳質材料是一類能夠用于去除水體中有機污染物的催化材料,表面具有豐富的活性位點,可以活化多種氧化劑. Sun 等研究發(fā)現,碳納米管(CNT)可以在較寬pH 范圍(7.0~10.0)內加速Fe(Ⅵ)降解溴苯酚類化合物,在pH=8.0 時,加入25、50、100 mg/L 的CNT 可將3-溴苯酚降解的準一級速率常數由Fe(Ⅵ)直接氧化體系的0.003 4 s分別升至0.005 9、0.011、0.016 s,推測活化機理為CNT 表面酚羥基與Fe(Ⅵ)反應生成高活性的Fe(Ⅳ)/Fe(Ⅴ). Fe(Ⅵ)/CNT體系能夠選擇性氧化富電子有機污染物(如溴苯酚類化合物、磺胺甲惡唑等),但對少電子的有機污染物(如碘帕醇、硝基苯等)的反應活性低,體現了Fe(Ⅳ)/Fe(Ⅴ)的氧化選擇性. Pan 等進一步比較研究了多種碳質材料對Fe(Ⅵ)的活化能力,發(fā)現碳質材料本身不吸附或少量吸附有機污染物,但可以通過表面的羰基活化Fe(Ⅵ)生成Fe(Ⅳ)/Fe(Ⅴ),促進有機污染物的降解. 碳質材料的活化能力與其種類有關,水熱炭的活化能力最強. 但由于碳質材料本身也可能存在環(huán)境風險和毒性危害,因此后續(xù)仍需對碳質材料活化Fe(Ⅵ)技術進行安全風險評估. 生物炭是環(huán)境友好的碳質材料,是由生物質在厭氧條件下熱解形成的,具有成本低、表面含氧官能團豐富等優(yōu)點,并且具有良好的催化性能. Tian 等研究發(fā)現,生物炭也可以活化Fe(Ⅵ)有效降解有機污染物,加入生物炭后,Fe(Ⅵ)對5 種有機污染物(磺胺甲惡唑、卡馬西平、環(huán)丙沙星、雙氯芬酸、N,N-二乙基-3-甲基苯甲酰胺)的氧化速率提高了3~14 倍,TOC 的去除率增加了2.4~8.0 倍,因此生物炭活化Fe(Ⅵ)技術是水處理和污水處理中有潛力的氧化方法. 通過甲基苯基亞砜探針化合物的試驗表明,Fe(Ⅳ)/Fe(Ⅴ)是起作用的活性物種,推測機理為生物炭表面的供電子基團(如酚羥基)將電子轉移給Fe(Ⅵ),活化Fe(Ⅵ)生成高活性的Fe(Ⅳ)/Fe(Ⅴ),從而促進有機污染物的降解.
綜上,Fe(Ⅵ)的異相活化技術主要是通過固體活化劑的表面作用或緩釋活化劑將Fe(Ⅵ)活化為高活性的Fe(Ⅳ)/Fe(Ⅴ). 因此,可以尋找具有特定官能團或具有緩釋能力的固體活化劑,開發(fā)新型異相活化技術. Fe(Ⅵ)的活化技術具有受到基質影響小、反應速率快等優(yōu)點,其機理如圖3 所示. 但對于某些Fe(Ⅵ)難以氧化的有機污染物,活化后可能也難以去除. 而Fe(Ⅵ)協(xié)同技術可以通過產生高活性的自由基(如OH、SO)等活性物種,有效去除與Fe(Ⅵ)反應活性低的有機污染物.

圖 3 Fe(Ⅵ)活化技術機理示意Fig.3 Diagram of the mechanisms of Fe(Ⅵ)activation technologies
3 Fe(Ⅵ)協(xié)同技術
Fe(Ⅵ)的協(xié)同技術通過將Fe(Ⅵ)氧化法和紫外光或協(xié)同劑相結合,實現對有機污染物的協(xié)同去除.相比于Fe(Ⅵ)活化技術,Fe(Ⅵ)協(xié)同技術可能會產生Fe(Ⅳ)/Fe(Ⅴ)之外的新的活性物種,如多種活性自由基(OH、SO、O)等. 與Fe(Ⅵ)的活化技術不同,Fe(Ⅵ)協(xié)同技術不局限于中性及偏堿性條件,在酸性條件下也可以實現Fe(Ⅵ)的協(xié)同作用. Fe(Ⅵ)的協(xié)同技術主要包括Fe(Ⅵ)/氧化劑協(xié)同、Fe(Ⅵ)/含硫還原劑協(xié)同、Fe(Ⅵ)/紫外光協(xié)同和Fe(Ⅵ)/光催化協(xié)同.
3.1 Fe(Ⅵ)/氧化劑協(xié)同
Fe(Ⅵ)可與多種常用水處理氧化劑〔如過硫酸氫鹽(PMS)、過硫酸鹽(PDS)等〕產生協(xié)同作用,促進有機污染物的降解,協(xié)同作用的機理為Fe(Ⅵ)的還原產物與氧化劑反應生成活性自由基 (如OH、SO).Feng 等使用Fe(Ⅵ)/PMS 體系降解4 種氟喹諾酮類化合物(FQs),發(fā)現向Fe(Ⅵ)和有機污染物混合體系中投加PMS 后,相比于Fe(Ⅵ)直接氧化,氟甲喹、恩諾沙星、馬波沙星、氧氟沙星的降解率分別提高了42%、15%、24%和28%. PMS 可以將Fe(Ⅵ)的還原產物Fe(Ⅲ)還原為Fe(Ⅱ),之后Fe(Ⅱ)催化PMS產生SO,促進FQs 的降解. Wu 等使用Fe(Ⅵ)/PMS體系氧化降解阿特拉津,發(fā)現Fe(Ⅵ)/PMS 體系能夠在較寬pH 范圍(5.0~9.0)內有效去除阿特拉津. pH=6.0 時,Fe(Ⅵ)直接氧化體系和Fe(Ⅵ)/PMS 體系中阿特拉津的降解率分別為11.7%和81.5%. 電子順磁共振結果和自由基抑制試驗表明,Fe(Ⅵ)/PMS 體系可以產生OH 和SO,而SO是導致阿特拉津降解的主要活性物種. 此外,除Fe(Ⅲ)外,Fe(Ⅵ)的還原產物氧化鐵(FeO)也可以活化PMS 生成SO,促進阿特拉津的降解. Li 等發(fā)現,Fe(Ⅵ)/PDS 體系可以協(xié)同降解抗生素環(huán)丙沙星,投加羥胺可以加速Fe(Ⅲ)的還原和Fe(Ⅱ)的再生,從而進一步提高Fe(Ⅵ)/PDS體系對環(huán)丙沙星的降解率. Fe(Ⅵ)直接氧化體系、Fe(Ⅵ)/PDS 體系和Fe(Ⅵ)/PDS/羥胺體系中環(huán)丙沙星的降解率分別為54.2%、72.6%、91.5%,Fe(Ⅵ)及產生的OH 和SO是Fe(Ⅵ)/PDS 體系的主要活性物種.
3.2 Fe(Ⅵ)/含硫還原劑協(xié)同
亞硫酸鹽(SO)是優(yōu)良的還原劑,Feng 等研究發(fā)現,Fe(Ⅵ)/SO體系可以在15 s 內提高難降解抗生素甲氧芐啶及氟甲喹的降解率,并推測在該體系中,多種活性物種〔Fe(Ⅳ)/Fe(Ⅴ)、SO、SO和OH〕可能參與氧化反應,有機污染物的降解率取決于溶液pH 和[SO]/[Fe(Ⅵ)] (濃 度 比). Sun 等研 究發(fā)現,Fe(Ⅵ)/SO體系可以在10 s 內降解約78%的N,N-二乙基-3-甲苯酰胺,而Fe(Ⅵ)直接氧化不發(fā)生降解,并確認SO為主要活性物種. 而Zhang 等使用Fe(Ⅵ)/SO2氧化降解磺胺甲惡唑,發(fā)現OH 和SO是主要活性物種. 為了準確鑒定此體系中起作用的活性物種,Shao 等選取難溶的CaSO作為SO的緩釋源,研究了Fe(Ⅵ)-CaSO體系的氧化效能并對活性物種進行了鑒定,發(fā)現Fe(Ⅵ)-CaSO體系的氧化機理主要是溶解的SO2通過單電子轉移直接將Fe(Ⅵ)還原為Fe(Ⅳ)/Fe(Ⅴ). 該體系可以有效氧化多種有機污染物(磺胺甲惡唑、恩諾沙星、卡馬西平、雙氯芬酸鈉、阿特拉津、布洛芬和苯甲酸),氧化速率比Fe(Ⅵ)直接氧化快6.1~173.7 倍,且對實際水體中的有機污染物也有較好的去除效果. 綜上,Fe(Ⅵ)/SO2體系雖然具有快速去除有機污染物的潛力,但不同研究中主要活性物種不同,這可能是由試驗條件不同所致,如SO2與Fe(Ⅵ)濃度比的變化可使活性物種發(fā)生變化. Shao 等研究發(fā)現,在Fe(Ⅵ)/SO2體系中,活性物種的生成情況與SO2與Fe(Ⅵ)的濃度比相關. 當SO2與Fe(Ⅵ)的濃度比為0.1~0.3 時,Fe(Ⅴ)為主要活性物種;當SO2與Fe(Ⅵ)的濃度比為0.4~1.0 時,Fe(Ⅴ)、OH 和SO為主要活性物種;當SO2與Fe(Ⅵ)的濃度比為1.5~10.0 時,OH 和SO為主要活性物種. 此外,相比于單次投加,分次投加SO2可以進一步提高有機污染物的去除效能.
除SO2外,硫代硫酸鹽(SO)也可以協(xié)同Fe(Ⅵ)促進有機污染物降解,但其活性物種存在爭議. Zhang等將Fe(Ⅵ)與SO2結合,發(fā)現該體系能夠協(xié)同降解氯霉素,主要氧化活性物種為OH 和SO的作用,而Gao 等研究結果表明Fe(Ⅵ)/SO2體系起作用的活性物種只有Fe(Ⅳ)/Fe(Ⅴ). 因此,對于Fe(Ⅵ)/SO體系的活性物種仍需進一步探究.
3.3 Fe(Ⅵ)/紫外光協(xié)同
紫外照射(UV)是水處理中常用的手段,Aslani等發(fā)現Fe(Ⅵ)/UV 體系能夠協(xié)同去除鹵乙酸,推測Fe(Ⅵ)、Fe(Ⅴ)、OH 為主要活性物種. 而Wu 等使用Fe(Ⅵ)/UV 體系降解2,4-二氯苯酚,發(fā)現Fe(Ⅵ)/UV體系降解2,4-二氯苯酚的速率常數分別比UV 體系和Fe(Ⅵ)氧化體系高6.9 和9.2 倍,O為主要活性物種. 張雄軍等使用UV 和Fe(Ⅵ)結合降解雙酚A,發(fā)現Fe(Ⅵ)/UV 體系具有明顯協(xié)同作用,與Fe(Ⅵ)直接氧化相比,協(xié)同體系的COD去除率提高了15%左右(UV 單獨作用時去除率為1.5%). 該體系的協(xié)同作用是由于UV 提高了雙酚A 的反應活性,使其更易于被Fe(Ⅵ)降解,并且Fe(Ⅵ)在自分解過程中產生的HO和UV 構成了UV/HO高級氧化體系,從而促進雙酚A 的降解. 綜上,Fe(Ⅵ)/UV 體系的機理存在爭議,仍需進一步探究.
3.4 Fe(Ⅵ)/光催化協(xié)同
Fe(Ⅵ)/光催化體系研究主要集中于Fe(Ⅵ)/UV/TiO體系. Fe(Ⅵ)和光催化的協(xié)同氧化是利用TiO光催化(UV/TiO)產生的導帶電子還原Fe(Ⅵ),得到高活性的Fe(Ⅴ). 由于Fe(Ⅵ)接收了導帶電子,減少了導帶電子和空穴的復合,從而提高空穴的氧化效率. 空穴協(xié)同Fe(Ⅴ)、Fe(Ⅵ)氧化水中的有機污染物,從而實現協(xié)同效果. Yuan 等將Fe(Ⅵ)與UV/TiO光催化體系相結合,發(fā)現Fe(Ⅵ)/UV/TiO體系可以協(xié)同降解鄰苯二甲酸二甲酯. pH=9.0 時,鄰苯二甲酸二甲酯在Fe(Ⅵ)直接氧化體系中幾乎不降解,在UV/TiO體系中的降解率為12%,而在Fe(Ⅵ)/UV/TiO體系中的降解率達到了65%,體現了明顯的協(xié)同作用.
綜上,Fe(Ⅵ)的活化技術是通過生成高活性的Fe(Ⅳ)/Fe(Ⅴ)對Fe(Ⅵ)直接氧化進行強化,而Fe(Ⅵ)的協(xié)同技術則是通過生成活性自由基(如OH、SO、O等)對Fe(Ⅵ)直接氧化進行強化,協(xié)同技術的機理如圖4 所示. Fe(Ⅵ)活化及協(xié)同技術可以提高Fe(Ⅵ)對有機污染物的降解率及降解速率,降低Fe(Ⅵ)用量,去除與Fe(Ⅵ)反應活性低的有機污染物,因此Fe(Ⅵ)活化及協(xié)同技術既可以保留Fe(Ⅵ)氧化不易受到環(huán)境基質影響、產物具有吸附絮凝能力的優(yōu)點,還能夠通過生成高活性的氧化物種提高Fe(Ⅵ)的氧化能力與氧化容量,加快有機污染物的降解并提高其降解率. Fe(Ⅵ)的活化技術可以降低環(huán)境基質對Fe(Ⅵ)的影響,不足之處是Fe(Ⅵ)活化產生的Fe(Ⅳ)/Fe(Ⅴ)可能也難以氧化與Fe(Ⅵ)反應活性低的有機污染物,因為Fe(Ⅳ)/Fe(Ⅴ)對有機污染物的氧化機理與Fe(Ⅵ)類似,都是通過電子轉移進行的.Fe(Ⅵ)協(xié)同技術可以生成高活性的自由基(如OH、SO等),氧化Fe(Ⅵ)不能直接降解的有機污染物,不足之處是活性自由基的低選擇性會導致其受到環(huán)境基質的影響較大. 因此,需要根據目標有機污染物種類和水體基質成分選擇合適的Fe(Ⅵ)活化或協(xié)同技術進行處理. Fe(Ⅵ)的活化及協(xié)同技術對有機污染物降解的促進效果與活性物種見表1.

圖 4 Fe(Ⅵ)協(xié)同技術機理示意Fig.4 Diagram of the mechanisms of Fe(Ⅵ)synergy technologies
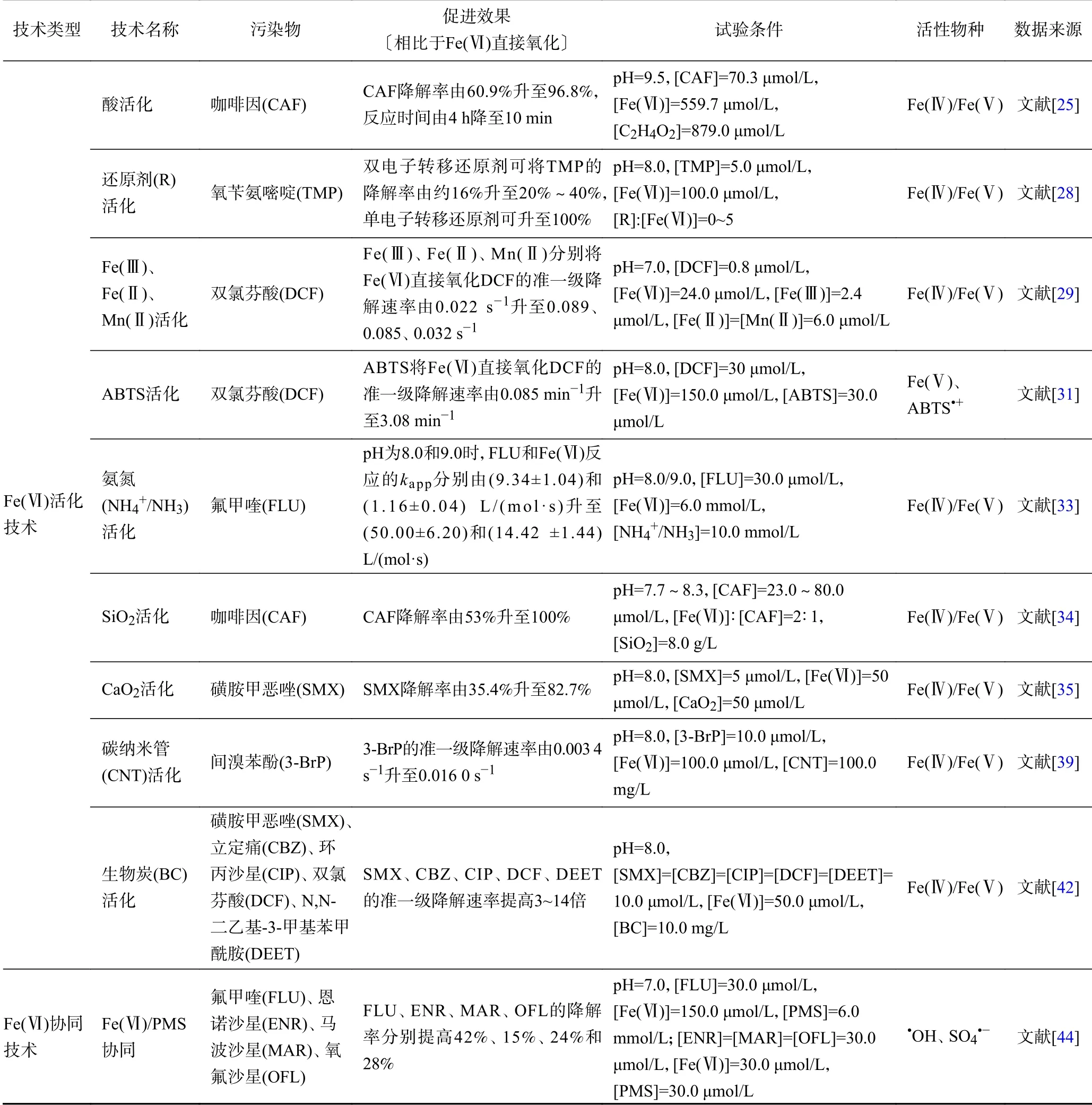
表 1 Fe(Ⅵ)活化及協(xié)同技術對有機污染物降解的促進效果與活性物種Table 1 The promotion effect on the degradation of organic pollutants and active species by Fe(Ⅵ) activation and synergy technologies
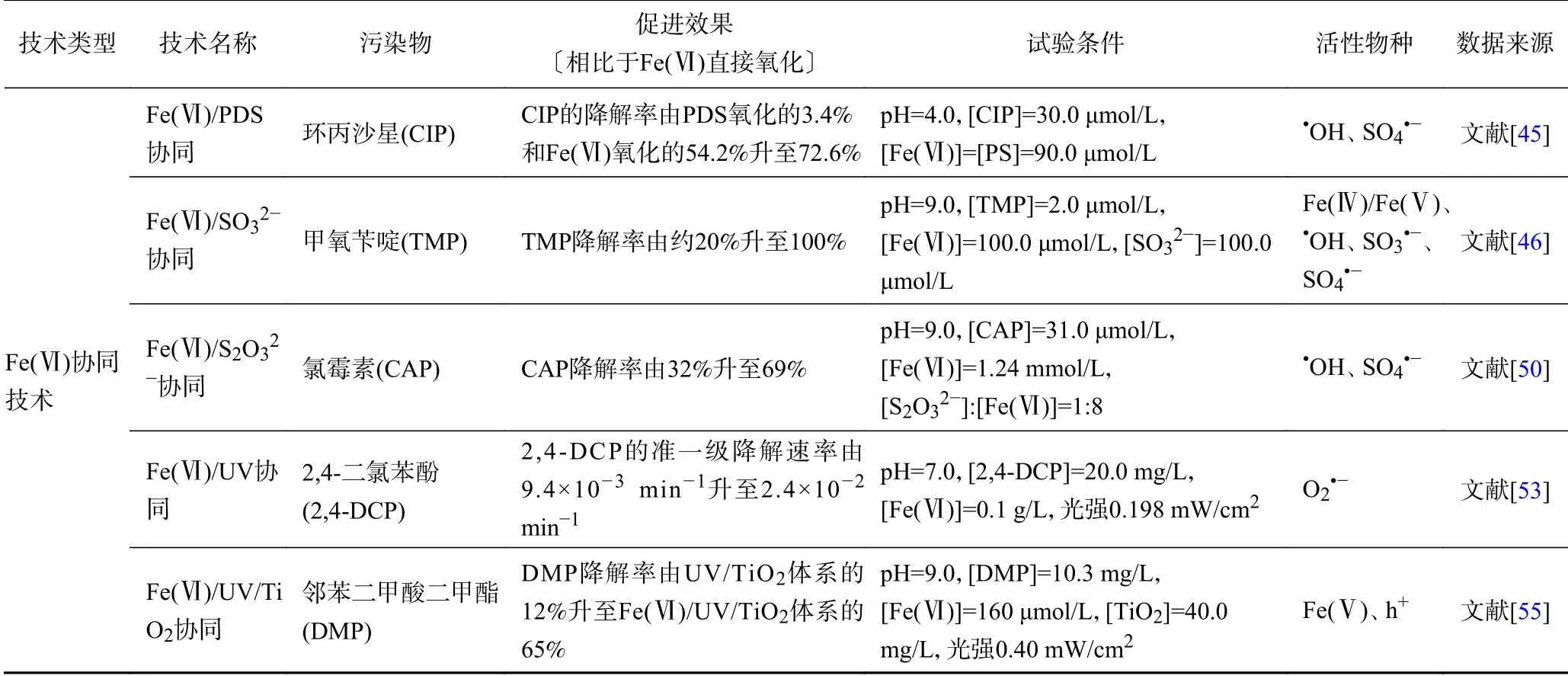
續(xù)表 1
4 Fe(Ⅵ)氧化的強化技術對有機污染物降解產物種類、分布及毒性影響
同種有機污染物經過不同技術處理,其降解產物種類、分布及毒性都可能發(fā)生變化. 由于Fe(Ⅵ)活化及協(xié)同技術是通過產生新的活性物種對有機污染物進行降解,因此降解機理、降解產物的種類、分布和毒性等都可能有所變化. 現有的Fe(Ⅵ)活化及協(xié)同技術更多關注于有機污染物的降解效能及Fe(Ⅵ)活化或協(xié)同技術的機理探究,對有機污染物的降解產物及其毒性變化研究較少.
相比于Fe(Ⅵ)直接氧化法,Fe(Ⅵ)的活化技術可生成高活性的Fe(Ⅳ)/Fe(Ⅴ). Fe(Ⅳ)/Fe(Ⅴ)與Fe(Ⅵ)氧化有機污染物的機理均為電子轉移,但其自身性質可能不同于Fe(Ⅵ),從而改變有機污染物的降解產物. Feng 等使用氨氮活化Fe(Ⅵ)氧化降解氟甲喹時發(fā)現,與Fe(Ⅵ)直接氧化相比,氟甲喹降解產物的種類不變,但產物分布發(fā)生變化. 這是因為氨氮絡合的Fe(Ⅳ)/Fe(Ⅴ)更易于進攻氟甲喹的C=C鍵,從而導致氨氮存在時,碳碳雙鍵斷裂的產物在液相色譜-質譜聯(lián)用儀中測定的峰面積明顯升高,而相應的羥基化產物的峰面積明顯下降. Fe(Ⅵ)協(xié)同技術是通過產生新的活性物種(如活性自由基等)促進有機污染物的降解,Fe(Ⅵ)與協(xié)同試劑的濃度比及投加方式均可能對有機污染物的降解產物帶來影響. Shao 等發(fā)現,使用Fe(Ⅵ)/SO2協(xié)同體系降解恩諾沙星的產物與SO與Fe(Ⅵ)的濃度比有關,在不同濃度比下,有不同產物生成. Gong 等將紫外光與Fe(Ⅵ)/PMS協(xié)同體系相結合降解磺胺甲惡唑,發(fā)現Fe(Ⅵ)、OH和SO是主要的活性物種,且不同試劑投加方式對磺胺甲惡唑降解產物的種類和分布有所影響. 單次投加Fe(Ⅵ)時,磺胺甲惡唑的主要降解路徑為硝基化、羧基化、脫甲基、羥基化和苯環(huán)的開環(huán);分步投加Fe(Ⅵ)時,磺胺甲惡唑的主要降解路徑為羥基化、羧基化和S—N 鍵的斷裂;而分步投加PMS 時,磺胺甲惡唑的主要降解路徑為羥基化和羧基化. 這是由于不同試劑投加方式可能導致活性物種的比例發(fā)生變化,從而改變磺胺甲惡唑降解產物的種類及分布. 相比于Fe(Ⅵ)直接氧化法,Fe(Ⅵ)協(xié)同體系生成的自由基(如OH、SO)氧化能力更強,且選擇性低,因此可能生成更多Fe(Ⅵ)直接氧化不能生成的降解產物. 因此,Fe(Ⅵ)協(xié)同體系除Fe(Ⅵ)直接氧化的降解途徑以外,還會有新的降解途徑,降解產物種類總體是增加的. 例如,Feng 等使用Fe(Ⅵ)/PMS 協(xié)同體系降解氟甲喹,發(fā)現相比于Fe(Ⅵ)直接氧化,協(xié)同體系檢測到了3 種脫氟產物,可能是SO作用導致的降解路徑.
目前對于Fe(Ⅵ)活化前后反應體系毒性對比的相關研究十分有限. Cao 等使用Fe-苯酚改性生物炭活化Fe(Ⅵ)降解阿特拉津,通過發(fā)光細菌急性毒性測試評估反應前后溶液毒性的變化,反應30 min后,相比于阿特拉津原始溶液,Fe(Ⅵ)直接氧化體系的抑制率降低了11.38%,Fe-苯酚改性生物炭活化Fe(Ⅵ)體系的抑制率降低了19.02%,表明Fe-苯酚改性生物炭活化Fe(Ⅵ)體系對阿特拉津溶液毒性降低效果更明顯. Zhang 等使用CaO緩釋HO,以活化Fe(Ⅵ)降解磺胺甲惡唑,發(fā)現反應結束后,磺胺甲惡唑的原始溶液、Fe(Ⅵ)氧化體系、CaO活化Fe(Ⅵ)體系的抑制率分別為81.66%、68.78%和71.36%,表明CaO活化Fe(Ⅵ)體系與未活化體系反應后的毒性接近,兩種體系均能有效降低磺胺甲惡唑溶液的毒性.
Fe(Ⅵ)活化及協(xié)同技術可以導致活性物種發(fā)生變化,從而影響有機污染物的降解產物種類和分布,進而影響反應體系的毒性. 研究Fe(Ⅵ)活化及協(xié)同技術氧化有機污染物的產物及毒性變化情況對評價該技術是否適用于實際水體處理十分重要,但目前有關Fe(Ⅵ)活化及協(xié)同技術降解有機污染物的毒性變化研究十分有限,需進一步加強相關研究.
5 結論與展望
a) 在Fe(Ⅵ)活化體系中,Fe(Ⅵ)轉化為高活性的Fe(Ⅳ)/Fe(Ⅴ),實現對有機污染物的快速氧化. 但一方面,均相活化體系的活化劑濃度越高,產生Fe(Ⅳ)/Fe(Ⅴ)的速率越快,Fe(Ⅳ)/Fe(Ⅴ)的自分解也越劇烈;另一方面,使用還原劑活化Fe(Ⅵ)時,過量的還原劑會競爭消耗產生的Fe(Ⅳ)/Fe(Ⅴ),從而降低活化效果,如HO活化Fe(Ⅵ)時,過量的HO會競爭消耗Fe(Ⅳ)/Fe(Ⅴ),而使用CaO緩釋HO可以提高Fe(Ⅵ)的活化效果. 因此通過具有緩釋能力的異相活化劑,如開發(fā)固體酸催化劑緩釋H以活化Fe(Ⅵ),可以減緩Fe(Ⅳ)/Fe(Ⅴ)的生成,抑制其自分解,減少還原劑對Fe(Ⅳ)/Fe(Ⅴ)的消耗,進而提高Fe(Ⅵ)活化技術去除有機污染物的效能.
b) Fe(Ⅵ)活化及協(xié)同技術能夠明顯提高Fe(Ⅵ)氧化有機污染物的效能,縮短反應時間. 但相關研究主要關注于對有機污染物降解的促進效果,部分技術〔如酸活化Fe(Ⅵ)技術〕的機理未知或存在爭議,需對Fe(Ⅵ)的活化及協(xié)同機理進行進一步探究.
c) Fe(Ⅵ)活化及協(xié)同技術可以生成新的活性物種,從而對有機污染物降解產物的種類、分布及毒性產生影響. 目前對有機污染物降解產物變化的相關研究較少,需深入探究Fe(Ⅵ)活化及協(xié)同技術對有機污染物降解產物種類、分布及毒性的影響.
d) Fe(Ⅵ)活化及協(xié)同技術的相關研究主要在純水體系中進行,對于實際廢水中的有機污染物去除研究較少. 實際廢水基質復雜,有機污染物種類多、濃度低,未來應加強對實際廢水中有機污染物的處理效能研究.
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
汽車觀察(2018年10期)2018-11-06 07:05:26
少兒科學周刊·少年版(2015年1期)2015-07-07 17:15:12
現代企業(yè)(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
