烏藥湯揮發性成分提取工藝的優化
2022-03-29 23:42:29程媛孫吳倩劉明松鄧亞偉段續紅李春花
中國藥房 2022年6期
程媛 孫吳倩 劉明松 鄧亞偉 段續紅 李春花

中圖分類號 R282 文獻標志碼 A 文章編號 1001-0408(2022)06-0712-06
DOI 10.6039/j.issn.1001-0408.2022.06.11
摘 要 目的 優化經典名方烏藥湯揮發性成分的提取工藝。方法 在單因素實驗的基礎上,以有效成分乙酸龍腦酯、香附烯酮、α-香附酮、藁本內酯、去氫木香內酯含量及揮發油提取率的綜合評分為指標,以提取時間、浸泡時間、液料比為響應因素,采用Box-Behnken設計-響應面法優化烏藥湯揮發性成分的提取工藝并驗證。在此基礎上,再對藥液的提取狀態進行量化。結果 最優提取工藝為液料比13 ∶ 1(mL/g),浸泡時間0.5 h,暴沸狀態下提取6 h。3次驗證實驗的綜合評分分別為0.948 7、0.948 4、0.948 6(RSD=0.02%,n=3),與預測值(0.947 9)的偏差均不超過1%。以180 ℃油浴下藥液的沸騰狀態作為暴沸狀態。結論 所得烏藥湯揮發性成分最優提取工藝穩定、可行。
關鍵詞 烏藥湯;揮發性成分;提取工藝;Box-Behnken設計-響應面法
Optimization of the extraction technology of volatile components from Wuyao decoction
CHENG Yuan1,SUN Wuqian1,LIU Mingsong1,DENG Yawei1,DUAN Xuhong1,LI Chunhua1,2,3(1. College of Pharmacy, Hebei University of Traditional Chinese Medicine, Shijiazhuang 050090, China; 2. Hebei Higher Education Institute Applied Technology Research Center on TCM Formula Preparation/Hebei Technology Innovation Center of TCM Formula Preparation, Shijiazhuang 050090, China; 3. Hebei Industrial Technology Institute for Traditional Chinese Medicine Preparation, Shijiazhuang 050033, China)
ABSTRACT? ?OBJECTIVE To optimize the extraction technology of volatile components from Wuyao decoction. METHODS On the basis of single factor investigation, the extraction technology of volatile components from Wuyao decoction was optimized and validated by Box-Behnken design-response surface technology using the contents of bomyl acetate, cyperotundone, α-cyperone, ligustilide and dehydrocostuslactone, extraction rate of volatile oil as indexes, with extraction time, soaking time and liquid-material ratio as factors. On this basis, the extraction state of the decoction was quantified. RESULTS The optimal extraction technology was as followed: the ratio of liquid-material was 13 ∶ 1 (mL/g), soaking time was 0.5 h, and the extraction time was 6 h in the boiling state. The comprehensive scores of the three validation experiments were 0.948 7, 0.948 4 and 0.948 6 respectively (RSD=0.02%, n=3), and the deviation from the predicted value (0.947 9) was no more than 1%. The boiling state of the decoction in 180 ℃ oil bath was taken as the sudden boiling state. CONCLUSIONS The optimized extraction technology is stable and feasible.
KEYWORDS? ?Wuyao decoction; volatile components; extraction technology; Box-Behnken design-response surface technology
經典名方烏藥湯由烏藥、香附、木香、當歸、甘草5味藥組成,主治婦人血海疼痛[1]。現代研究表明,烏藥湯中的烏藥、香附、木香、當歸均含有揮發性成分,對全方活血疏經作用的貢獻較大[2-4]。傳統湯劑可保留藥材中揮發性成分的療效,但也有研究指出,烏藥湯水煎液中揮發性成分的含量較低,在制備成制劑的過程中,揮發性成分不斷損失,難以發揮傳統湯劑的療效[5]。因此,本課題組考慮將該類藥材單獨提取后將所得揮發性成分包合入藥,以最大程度地保留藥效。
揮發性成分的有效收集和保留對提高烏藥湯療效和推動其現代研究至關重要。目前,水蒸餾法是收集揮發性成分的常用方法,其設備簡單、成本低廉,是工業上最常用的提取方法[6]。本研究在單因素實驗的基礎上,以有效成分乙酸龍腦酯、香附烯酮、α-香附酮、藁本內酯、去氫木香內酯含量及揮發油提取率的綜合評分為指標,以提取時間、浸泡時間、液料比為因素,結合Box-Behnken設計-響應面法優化烏藥湯揮發性成分的提取工藝,旨在為該方的開發利用提供參考。
1 材料
1.1 主要儀器
本研究所用主要儀器包括7890B型氣相色譜儀(美國Agilent公司),SPH-300型氫氣發生器、SPB-3型全自動空氣源(北京中惠普分析技術研究所),LQ-A10002型電子天平(上海樂祺精密電子廠),98-1B型電子調溫電熱套(天津市泰斯特儀器有限公司),YP2002型電子分析天平(上海津平科學儀器有限公司),DF-101S型集熱式恒溫加熱磁力攪拌器(鞏義市予華儀器有限責任公司)等。
1.2 主要藥品與試劑
烏藥飲片(批號20010301)、香附飲片(批號200501)、當歸飲片(批號200601c155)、木香飲片(批號20100501)、甘草飲片(批號20092201)均購自河北安國藥材市場,經河北中醫學院侯芳潔副教授鑒定,分別為樟科植物烏藥Lindera aggregata(Sims) Kosterm.的干燥塊根、莎草科植物莎草Cyperus rotundus L.的干燥根莖、傘形科植物當歸Angelica sinensis(Oliv.) Diels的干燥根、菊科植物木香Aucklandia lappa Decne.的干燥根、豆科植物甘草Glycyrrhiza uralensis Fisch.的干燥根,均符合2020年版《中國藥典》(一部)標準[7]。
乙酸龍腦酯對照品(批號MUST-20081605,純度97%)購自中國科學院成都生物研究所;去氫木香內酯對照品(批號MUST-19101209,純度≥98%)、α-香附酮對照品(批號MUST-20052102,純度≥98%)均購自成都曼斯特生物科技有限公司;香附烯酮對照品(批號Z1705241,純度≥98%)購自成都草源康生物科技有限公司;藁本內酯對照品(批號RFS-G01001901005,純度≥98%)購自成都瑞芬斯生物科技有限公司;甲醇(色譜純)購自賽默飛世爾科技(中國)有限公司;其余試劑均為分析純或實驗室常用規格,水為超純水。
2 方法與結果
2.1 揮發油提取率的測定
按照2020年版《中國藥典》(四部)通則“2204揮發油測定法(甲法)”[8]提取揮發油:按處方比例稱取烏藥20 g,當歸、甘草、木香各10 g,香附40 g,粉碎為最粗粉,置于2 000 mL圓底燒瓶中,加入12倍量水浸泡0.5 h,加熱,暴沸提取6 h后停止加熱,靜置30 min,精密讀取烏藥湯揮發油提取量,計算提取率:提取率(%)=提取量(mL)/全粉質量(g)×100%。
2.2 乙酸龍腦酯、香附烯酮、α-香附酮、藁本內酯、去氫木香內酯的含量測定
參考文獻[9],采用氣相色譜法測定乙酸龍腦酯、香附烯酮、α-香附酮、藁本內酯、去氫木香內酯的含量。
2.2.1 色譜條件 色譜柱為Agilent HP-5毛細管柱(30 m×0.25 mm,0.25 μm);檢測器為氫火焰離子化檢測器,程序升溫(起始溫度為60 ℃;以10 ℃/min升溫至110 ℃,保持1 min;以1 ℃/min升溫至132 ℃,保持5 min;以0.2 ℃/min升溫至133 ℃,保持5 min;以0.2 ℃/min升溫至134 ℃,保持3 min;以5 ℃/min升溫至220 ℃);檢測器溫度為260 ℃;載氣為氮氣;進樣口溫度為250 ℃;進樣量為1 μL,分流比為25 ∶ 1。
2.2.2 混合對照品溶液的制備 分別稱取α-香附酮、香附烯酮、乙酸龍腦酯、去氫木香內酯、藁本內酯對照品,精密稱定,用甲醇制成質量濃度分別為1.125、3.360、0.954、0.369、0.900 mg/mL的混合對照品溶液。
2.2.3 供試品溶液的制備 按照“2.1”項下方法提取揮發油,以4 000 r/min離心6 min,上清液加入適量無水硫酸鈉干燥,得到不含水分的揮發油。取上述揮發油,用甲醇定容于10 mL量瓶中,精密量取2 mL置于5 mL量瓶中,再用甲醇定容,過0.22 μm微孔濾膜,即得供試品溶液。
2.2.4 系統適用性試驗 取混合對照品溶液、供試品溶液和空白對照溶液(甲醇),按照“2.2.1”項下色譜條件進樣測定,記錄色譜圖(圖1)。結果顯示,供試品溶液在對照品溶液的相應位置有相同的色譜峰,各色譜峰與相鄰色譜峰的分離度均大于1.5,空白對照溶液對測定無干擾(圖略),表明本方法系統適用性良好。
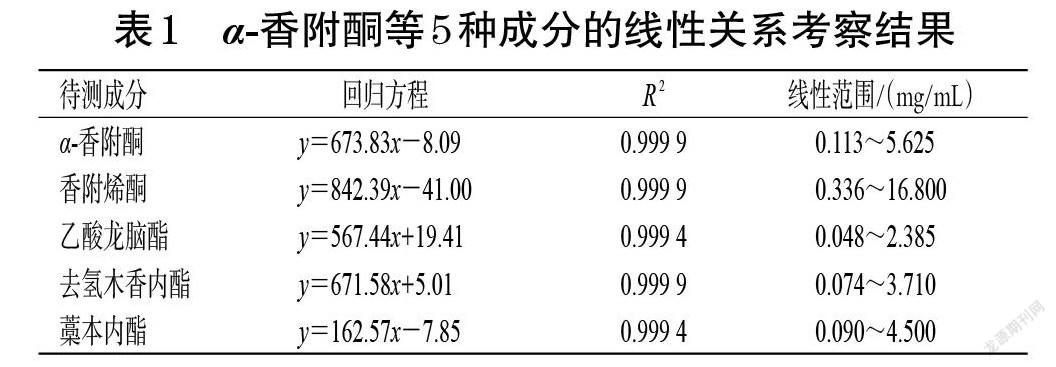
2.2.5 線性關系考察 稱取α-香附酮、香附烯酮、乙酸龍腦酯、去氫木香內酯、藁本內酯對照品適量,以甲醇配制成α-香附酮質量濃度分別為5.625、2.250、1.125、0.563、0.113 mg/mL,香附烯酮質量濃度分別為16.800、6.720、3.360、1.680、0.336 mg/mL,乙酸龍腦酯質量濃度分別為2.385、0.954、0.477、0.239、0.048 mg/mL,去氫木香內酯質量濃度分別為3.710、1.484、0.742、0.371、0.074 mg/mL,藁本內酯質量濃度分別為4.500、1.800、0.900、0.450、0.090 mg/mL的系列混合對照品溶液,按照“2.2.1”項下色譜條件進樣測定,記錄峰面積。以待測成分質量濃度(x)為橫坐標、峰面積(y)為縱坐標進行回歸分析,結果見表1。由表1可見,5種待測成分在各自線性范圍內均與峰面積成良好的線性關系。
2.2.6 精密度試驗 取“2.2.5”項下相應質量濃度(α-香附酮1.125 mg/mL、香附烯酮3.360 mg/mL、乙酸龍腦酯0.477 mg/mL、去氫木香內酯0.742 mg/mL、藁本內酯0.900 mg/mL)的混合對照品溶液適量,按照“2.2.1”項下色譜條件連續進樣測定6次,記錄峰面積。結果顯示,α-香附酮、香附烯酮、乙酸龍腦酯、去氫木香內酯、藁本內酯峰面積的RSD分別為0.97%、0.65%、0.35%、0.65%、0.31%(n=6),表明儀器精密度良好。
2.2.7 穩定性試驗 取供試品溶液適量,分別在室溫下放置0、2、4、8、12、24 h時按照“2.2.1”項下色譜條件進樣測定,記錄峰面積并代入回歸方程計算含量。結果顯示,α-香附酮、香附烯酮、乙酸龍腦酯、去氫木香內酯、藁本內酯含量的RSD分別為0.28%、0.27%、0.54%、0.41%、0.25%(n=6),表明供試品溶液在室溫下放置24 h內穩定性良好。
2.2.8 重復性試驗 按照“2.2.3”項下方法制備供試品溶液6份,按照“2.2.1”項下色譜條件進樣測定,記錄峰面積并代入回歸方程計算含量。結果顯示,α-香附酮、香附烯酮、乙酸龍腦酯、去氫木香內酯、藁本內酯含量的RSD分別為0.35%、0.45%、1.38%、0.38%、0.42%(n=6),表明本方法重復性良好。
2.2.9 加樣回收率考察 取已知待測成分含量的供試品溶液6份,精密加入一定量相應質量濃度(α-香附酮1.125 mg/mL、香附烯酮3.360 mg/mL、乙酸龍腦酯0.477 mg/mL、去氫木香內酯0.742 mg/mL、藁本內酯0.900 mg/mL)的混合對照品溶液,按照“2.2.3”項下方法制備供試品溶液,按照“2.2.1”項下色譜條件進樣測定,記錄峰面積并計算加樣回收率。結果顯示,α-香附酮、香附烯酮、乙酸龍腦酯、去氫木香內酯、藁本內酯的平均加樣回收率分別為98.75%、98.21%、98.41%、98.19%、99.79%,RSD均小于3%(n=6),表明本方法準確度好。
2.3 單因素實驗初步篩選揮發性成分提取工藝
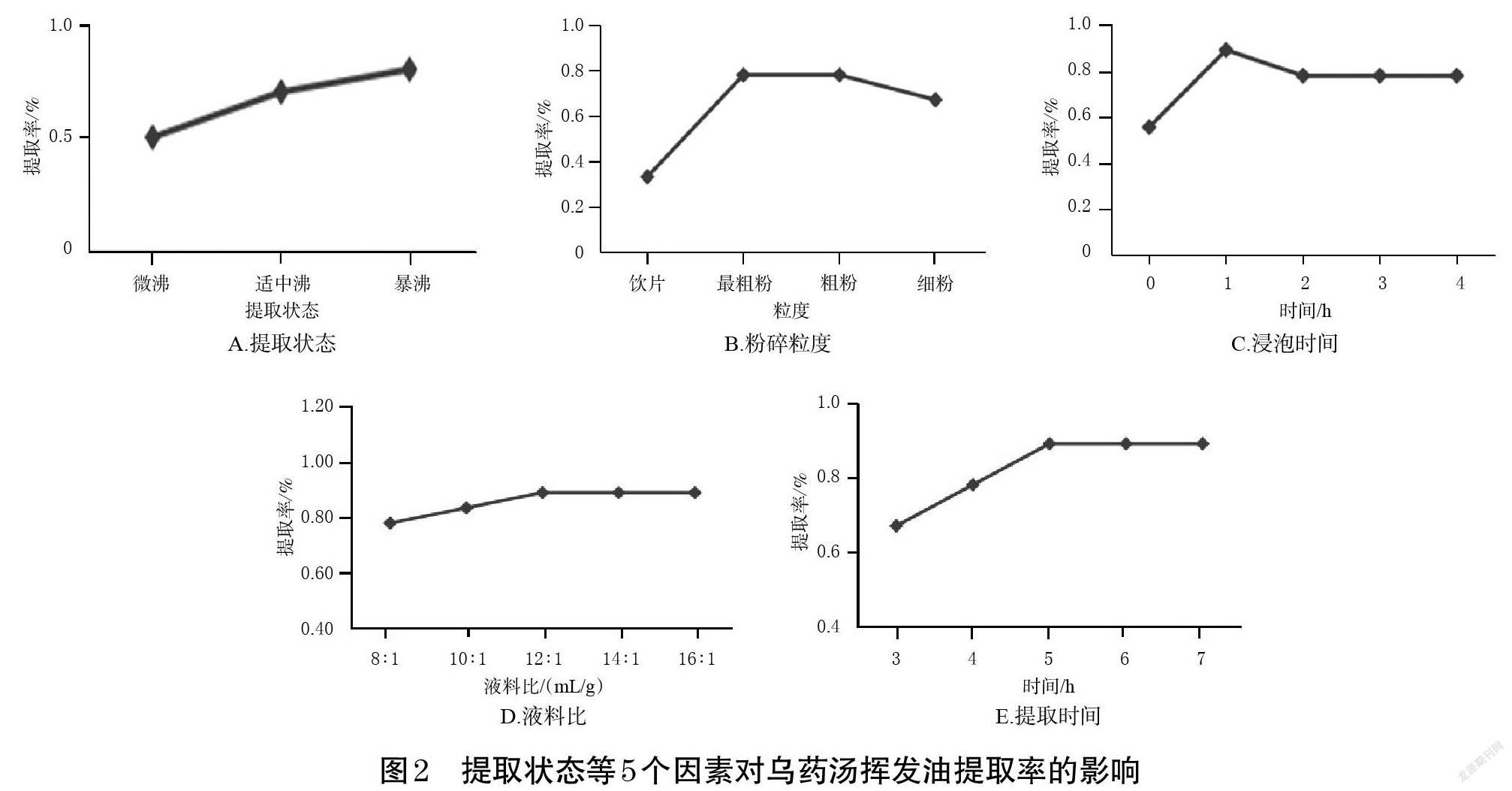
參考文獻[10],本課題組通過初步篩選影響烏藥湯揮發油提取率的因素發現,提取狀態、粉碎粒度、浸泡時間、液料比、提取時間5個因素對烏藥湯揮發油提取率的影響較大。因此,本研究以揮發油提取率為指標,以提取狀態(微沸、適中沸、暴沸)、粉碎粒度(飲片、最粗粉、粗粉、細粉,參照2020年版《中國藥典》(一部)“凡例”要求[7])、浸泡時間(0、1.0、2.0、3.0、4.0 h)、液料比(8 ∶ 1、10 ∶ 1、12 ∶ 1、14 ∶ 1、16 ∶ 1,mL/g)、提取時間(3、4、5、6、7 h)為因素,在固定其余4個因素的條件下進行單因素實驗,結果見圖2。
2.3.1 提取狀態 圖2A顯示,在固定其他因素提取烏藥湯揮發油時,火力越大,揮發油提取率越高,故選擇暴沸作為提取狀態。
2.3.2 粉碎粒度 圖2B顯示,在固定其他因素提取烏藥湯揮發油時,隨著粉碎粒度逐漸變小,揮發油提取率呈先升高后穩定再下降的趨勢;當粒度為最粗粉時,揮發油提取率達到峰值。筆者推測,這可能是因為中藥材被粉碎后,其油室受到了破壞,有利于提高提取效率,但粉碎越細藥材越容易結塊,且越不利于蒸餾[6]。故選擇最粗粉作為粉碎粒度。
2.3.3 浸泡時間 圖2C顯示,在固定其他因素提取烏藥湯揮發油時,隨著浸泡時間延長,揮發油提取率呈先升高后下降再平穩的趨勢;當浸泡時間為1.0 h時,揮發油提取率達到峰值,故選擇1.0 h作為浸泡時間。
2.3.4 液料比 圖2D顯示,在固定其他因素提取烏藥湯揮發油時,隨著液料比增加,揮發油提取率呈先升高后平穩的趨勢;當液料比為12 ∶ 1(mL/g)時,揮發油提取率達到峰值,故選擇12 ∶ 1(mL/g)作為液料比。
2.3.5 提取時間 圖2E顯示,在固定其他因素提取烏藥湯揮發油時,隨著提取時間延長,揮發油提取率呈先增加后平穩的趨勢;當提取時間為5 h時,揮發油提取率達到峰值,故選擇5 h作為提取時間。
2.4 Box-Behnken設計-響應面法優化揮發性成分提取工藝
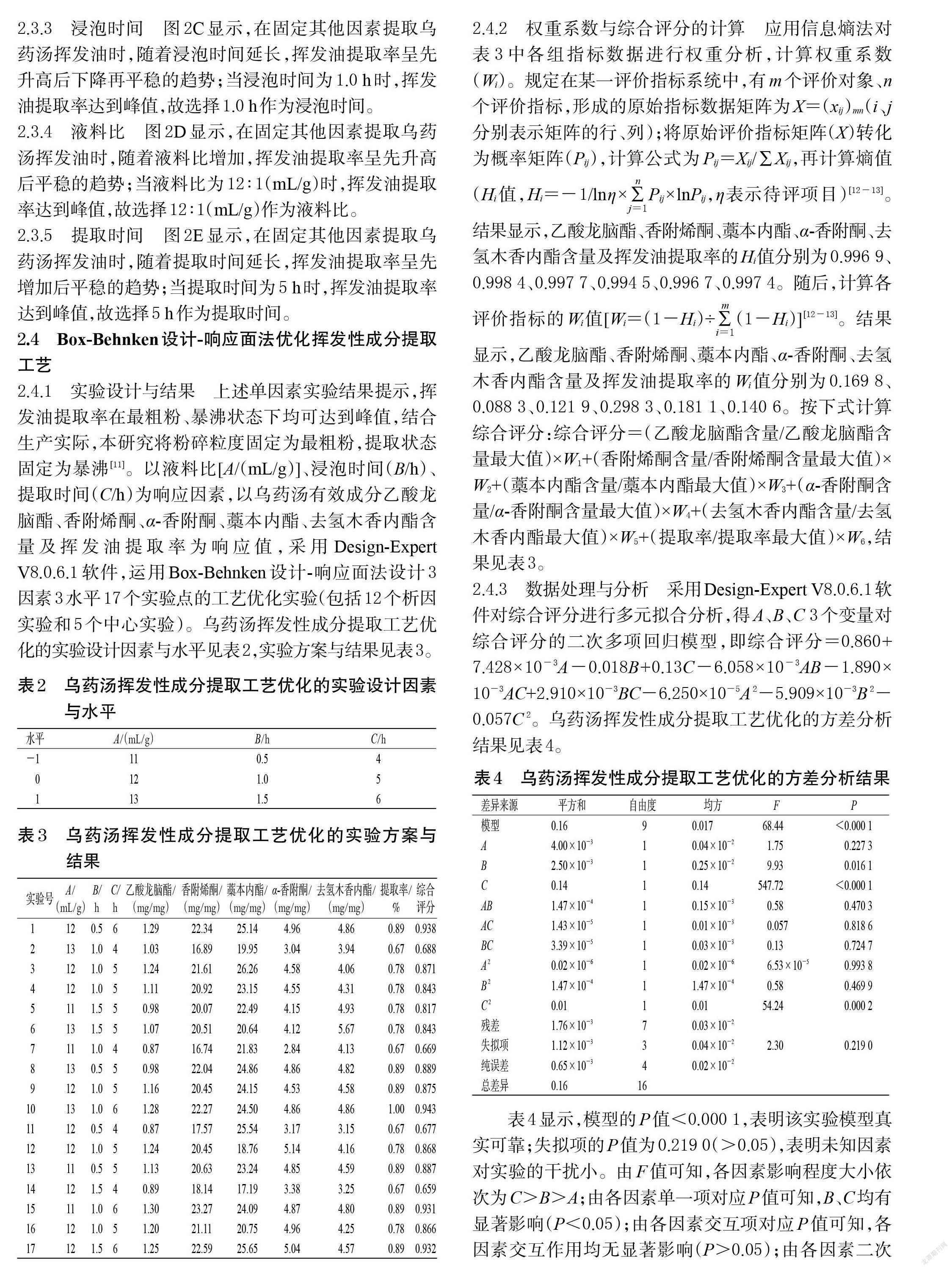
2.4.1 實驗設計與結果 上述單因素實驗結果提示,揮發油提取率在最粗粉、暴沸狀態下均可達到峰值,結合生產實際,本研究將粉碎粒度固定為最粗粉,提取狀態固定為暴沸[11]。以液料比[A/(mL/g)]、浸泡時間(B/h)、提取時間(C/h)為響應因素,以烏藥湯有效成分乙酸龍腦酯、香附烯酮、α-香附酮、藁本內酯、去氫木香內酯含量及揮發油提取率為響應值,采用Design-Expert V8.0.6.1軟件,運用Box-Behnken設計-響應面法設計3因素3水平17個實驗點的工藝優化實驗(包括12個析因實驗和5個中心實驗)。烏藥湯揮發性成分提取工藝優化的實驗設計因素與水平見表2,實驗方案與結果見表3。
2.4.2 權重系數與綜合評分的計算 應用信息熵法對表3中各組指標數據進行權重分析,計算權重系數(Wi)。規定在某一評價指標系統中,有m個評價對象、n個評價指標,形成的原始指標數據矩陣為X=(xij)mn(i、j分別表示矩陣的行、列);將原始評價指標矩陣(X)轉化為概率矩陣(Pij),計算公式為Pij=Xij/∑Xij,再計算熵值(Hi值,Hi=-1/lnη×[∑][j=1][n]Pij×lnPij,η表示待評項目)[12-13]。結果顯示,乙酸龍腦酯、香附烯酮、藁本內酯、α-香附酮、去氫木香內酯含量及揮發油提取率的Hi值分別為0.996 9、0.998 4、0.997 7、0.994 5、0.996 7、0.997 4。隨后,計算各評價指標的Wi值[Wi=(1-Hi)÷[∑][i=1][m](1-Hi)][12-13]。結果顯示,乙酸龍腦酯、香附烯酮、藁本內酯、α-香附酮、去氫木香內酯含量及揮發油提取率的Wi值分別為0.169 8、0.088 3、0.121 9、0.298 3、0.181 1、0.140 6。按下式計算綜合評分:綜合評分=(乙酸龍腦酯含量/乙酸龍腦酯含量最大值)×W1+(香附烯酮含量/香附烯酮含量最大值)×W2+(藁本內酯含量/藁本內酯最大值)×W3+(α-香附酮含量/α-香附酮含量最大值)×W4+(去氫木香內酯含量/去氫木香內酯最大值)×W5+(提取率/提取率最大值)×W6,結果見表3。
2.4.3 數據處理與分析 采用Design-Expert V8.0.6.1軟件對綜合評分進行多元擬合分析,得A、B、C 3個變量對綜合評分的二次多項回歸模型,即綜合評分=0.860+7.428×10-3A-0.018B+0.13C-6.058×10-3AB-1.890×10-3AC+2.910×10-3BC-6.250×10-5A 2-5.909×10-3B 2-0.057C 2。烏藥湯揮發性成分提取工藝優化的方差分析結果見表4。
表4顯示,模型的P值<0.000 1,表明該實驗模型真實可靠;失擬項的P值為0.219 0(>0.05),表明未知因素對實驗的干擾小。由F值可知,各因素影響程度大小依次為C>B>A;由各因素單一項對應P值可知,B、C均有顯著影響(P<0.05);由各因素交互項對應P值可知,各因素交互作用均無顯著影響(P>0.05);由各因素二次項對應P值可知,C 2有顯著影響(P<0.05);擬合方程的相關系數(R 2)為0.988 8,調整后的相關系數(R 2adj)為0.974 3,表明該模型可解釋97.43%響應值的變化,模型擬合程度較好、誤差較小。
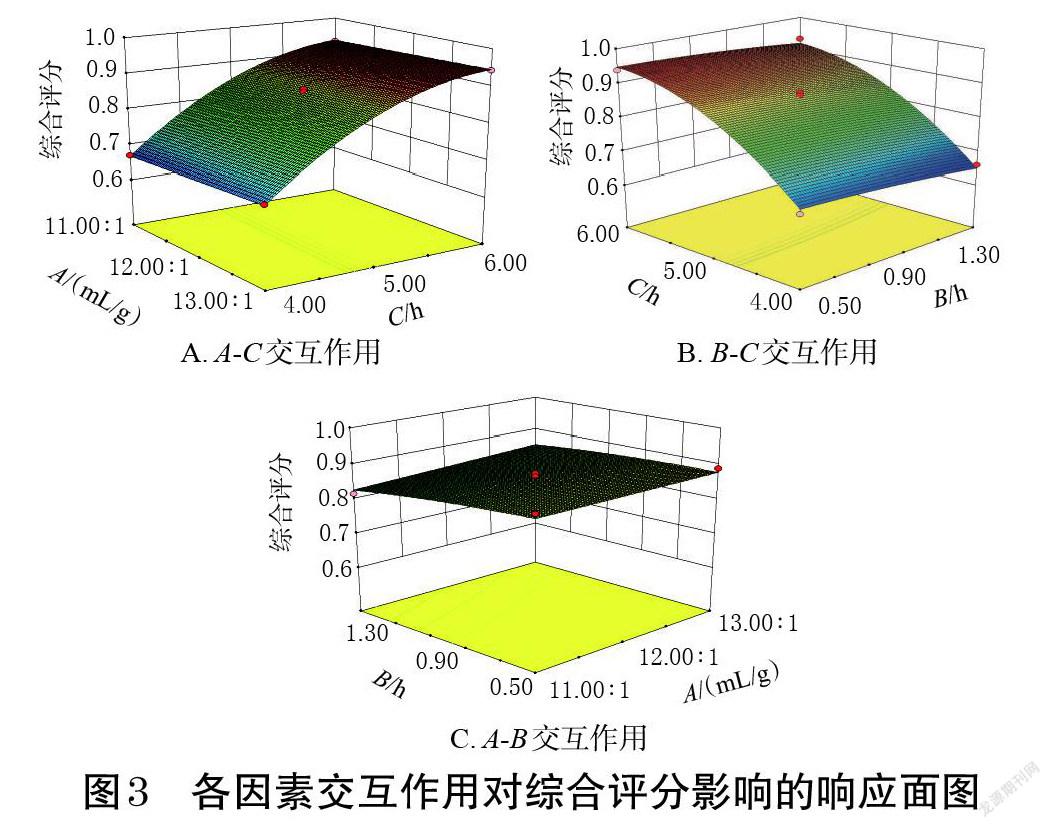
本研究采用Design-Expert V8.0.6.1軟件繪制各因素交互作用對綜合評分影響的響應面圖,結果見圖3。
響應面圖的陡峭程度可直接反映各因素交互作用的強弱[9]。圖3顯示,各因素交互作用的響應面圖均不陡峭,與表5中AB、AC、BC交互作用的影響不具統計學意義的結果一致。通過求解回歸模型方程得到最優提取工藝參數為:液料比12.99 ∶ 1(mL/g),浸泡時間0.62 h,提取時間6 h,綜合評分的預測值為0.947 9。結合生產實際,本研究對上述提取工藝參數進行修正,得到最優提取工藝參數為:藥材取最粗粉,液料比13 ∶ 1(mL/g),浸泡時間0.5 h,提取時間6 h。
2.4.4 驗證實驗 按照“2.4.3”項下最優提取工藝進行3次驗證實驗。結果顯示,綜合評分分別為0.948 7、0.948 4、0.948 6(RSD=0.02%,n=3),與預測值(0.947 9)的偏差均不超過1%,表明優化的提取工藝穩定、可行。
2.5 提取狀態的量化
參考文獻[14],按照“2.4.3”項下最優提取工藝進行實驗,采用油浴控溫裝置分別在120、150、170、180、190 ℃下提取烏藥湯揮發性成分,每個溫度條件平行操作3次,觀察6 h提取過程中藥液的沸騰狀態,并計算提取6 h時的提取率。結果顯示,在120 ℃下藥液沸騰狀態為微沸,平均提取率為0.56%;在150 ℃下藥液沸騰狀態為適中沸,平均提取率為0.72%;在170 ℃下藥液沸騰狀態為適中沸,平均提取率為0.78%;在180 ℃和190 ℃下藥液沸騰狀態為暴沸且180 ℃下的提取率更高,平均提取率為0.89%。故以180 ℃油浴下藥液的沸騰狀態作為暴沸狀態。
3 討論
本課題組預實驗發現,烏藥湯提取過程中烏藥、香附、木香、當歸、甘草揮發油不存在分層現象,表明處方中藥材揮發油密度相近,可同時提取。本研究采用Box-Behnken設計-響應面法與熵權法相結合的方式優化了烏藥湯揮發性成分的提取工藝。Box-Behnken設計-響應面法能綜合考察多個提取因素的影響,具有實驗次數少、精度高、預測值精準的優點;熵權法可在多指標篩選最優工藝時科學地賦予各指標相應的權重系數,以避免人為主觀因素的影響[12]。利用熵權法賦予各指標的權重系數計算綜合評分,再通過對綜合評分進行分析,從而篩選最優工藝,所得結果較為嚴謹[12-13,15]。
3.1 提取狀態的量化表征
外在的傳熱效率對揮發油提取的影響十分顯著,外在的加熱溫度或熱的傳遞效率會直接影響揮發油的蒸發量與蒸發速度,充分的蒸發量能保證揮發油提取完全[6]。中藥傳統理論多用“文火”“武火”表示,現多用“微沸”“暴沸”表示,均只能用操作者的主觀經驗來判斷,存在主觀性的弊端,因此尋找量化指標對提取狀態進行表征對工藝參數的完善有重要價值。本研究利用溫度對藥液提取狀態進行表征,結果顯示,在180 ℃和190 ℃油浴下提取狀態均為暴沸,但在180 ℃下提取率達到峰值,而在190 ℃下藥液沸騰過于劇烈,導致藥液不斷涌入揮發油收集器,不利于揮發油的收集。但是僅用溫度表征提取狀態還不夠全面,本課題組后期將考慮模擬實際大生產設備,深入研究可控參數,以達到提取狀態的精準化。
3.2 指標成分的選擇
在中藥產品的現代化工藝開發過程中,應基于有效成分的含量及其藥理活性進行綜合考慮。乙酸龍腦酯具有抗炎、鎮痛、調節胃腸運動、改善肝損傷的藥理作用,是烏藥的有效成分之一[16-18]。香附烯酮、α-香附酮存在于香附藥材中,成分特有性強,且在烏藥湯中的含量高[19-20]。藁本內酯是當歸揮發油的主要成分,具有解痙鎮痛、抗炎、緩解胃腸平滑肌痙攣的作用[21-22]。去氫木香內酯是木香的主要有效成分,且是2020年版《中國藥典》(一部)規定的含量指標成分。因此,本研究選擇乙酸龍腦酯、香附烯酮、α-香附酮、藁本內酯、去氫木香內酯作為評價烏藥湯的指標成分,并建立了測定其含量的氣相色譜法。方法學考察結果顯示,所建方法符合2020年版《中國藥典》(四部)的要求[8]。
綜上所述,本研究確定了經典名方烏藥湯揮發性成分的最優提取工藝,可為其進一步開發利用提供參考。
參考文獻
[ 1 ] 國家中醫藥管理局.關于發布《古代經典名方目錄(第一批)》的通知[EB/OL].[2021-09-21]. http://kjs.satcm.gov.cn/zhengcewenjian/2018-04-16/7107.html.
[ 2 ] 潘少斌,孔娜,李靜,等.香附化學成分及藥理作用研究進展[J].中國現代中藥,2019,21(10):1429-1434.
[ 3 ] 陳琳,朱靖,王嵩,等.當歸主要活性成分提取方法及其活性研究進展[J].上海醫藥,2021,42(9):71-75.
[ 4 ] 袁勝男,袁沖,毛志海,等.基于網絡藥理學對烏藥湯治療原發性痛經作用機制的研究[J].中國醫院藥學雜志,2020,40(10):1116-1121.
[ 5 ] 程媛,忻曉東,彭安堂,等.基于GC-MS指紋圖譜及多成分含量測定對經典名方烏藥湯物質基準質量評價研究[J].中國現代應用藥學,2021,38(23):2978-2984.
[ 6 ] 伍振峰,王賽君,楊明,等.中藥揮發油提取工藝與裝備現狀及問題分析[J].中國實驗方劑學雜志,2014,20(14):224-228.
[ 7 ] 國家藥典委員會.中華人民共和國藥典:一部[S]. 2020年版.北京:中國醫藥科技出版社,2020:57-241,ⅩⅥ .
[ 8 ] 國家藥典委員會.中華人民共和國藥典:四部[S]. 2020年版.北京:中國醫藥科技出版社,2020:233-234,480-483.
[ 9 ] 李佳佳,鄭鵬,頓佳穎,等.荊芥與連翹混合揮發油提取工藝優化[J].中國藥房,2019,30(6):813-817.
[10] 朱登輝,張靖柯,李孟,等. Box-Behnken設計-響應面法優化山茱萸葉總三萜提取工藝[J].中國藥房,2021,32(1):46-50.
[11] 張志瑞,李喜香,李盛華,等.祛寒逐風顆粒中揮發油提取工藝和包合工藝優化[J].中國藥房,2019,30(2):192- 196.
[12] 田帥,汪海濤,姜瑛,等.結合聚類分析與信息熵賦權的構件選擇方法[J].計算機與數字工程,2017,45(12):2437- 2441,2551.
[13] 陳云翔,董驍雄,項華春,等.基于信息熵的群組聚類組合賦權法[J].中國管理科學,2015,23(6):142-146.
[14] 武玉,周夢鴿,文甜甜,等.基于揮發油出油量的逍遙片提取工藝優化及揮發性成分研究[J].現代中藥研究與實踐,2019,33(6):40-45.
[15] 鄭鵬,李佳佳,頓佳穎,等.基于響應面法與信息熵法優選枳殼、醋香附總揮發性成分提取工藝[J].中國現代應用藥學,2019,36(12):1522-1528.
[16] 鄧桂明,向彪,肖小芹,等.基于網絡藥理學的烏藥主要化學成分藥效作用研究[J].中草藥,2018,49(21):5125- 5133.
[17] 邢夢雨,田崇梅,夏道宗.烏藥化學成分及藥理作用研究進展[J].天然產物研究與開發,2017,29(12):2147-2151.
[18] 晏潤緯,彭小梅.烏藥揮發油的化學成分及藥理作用[J].時珍國醫國藥,2014,25(11):2747-2749.
[19] 郭慧玲,駱云霞,胡律江,等.四制香附貼膏灸劑在大鼠體內的血液流變性與體外透皮特性分析[J].中國實驗方劑學雜志,2017,23(11):29-33.
[20] 胡棟寶,陸卓東,伍賢學.中藥香附子化學成分及藥理活性研究進展[J].時珍國醫國藥,2017,28(2):430-432.
[21] 柳小莉,黃小英,張小飛,等.基于GC-MS成分分析結合網絡藥理學預測當歸揮發油質量標志物(Q-Marker)[J].中草藥,2021,52(9):2696-2706.
[22] 吳國泰,王瑞瓊,杜麗東,等.當歸揮發油藥理作用研究進展[J].甘肅中醫藥大學學報,2018,35(4):87-92.
(收稿日期:2021-09-27 修回日期:2022-02-24)
(編輯:鄒麗娟)
