亨廷頓病一家系報道及相關(guān)文獻回顧分析
2022-03-09 13:09:54郭曉紅劉國榮武田琳許愛珍李秀娥
中風與神經(jīng)疾病雜志 2022年1期
張 元, 郭曉紅, 劉國榮, 吳 娟, 武田琳, 許愛珍, 李秀娥
亨廷頓病(Huntington disease,HD)又稱亨廷頓舞蹈病,臨床上以隱匿起病、緩慢進展的舞蹈癥、精神異常和癡呆為特征。該病是一種常染色體顯性遺傳性神經(jīng)變性病,遺傳方式為基因動態(tài)突變,具有遺傳早現(xiàn)特點。我們收集1例亨廷頓病家系,3代人中共有8例發(fā)病,本文對該家系先證者臨床癥狀及神經(jīng)影像學、基因檢測等結(jié)果進行分析總結(jié),并結(jié)合相關(guān)文獻進行討論。
1 資料和方法
1.1 臨床特點 先證者,女,41歲,已婚,無業(yè),初中畢業(yè),漢族。以“癲癇病”在外院治療無效,于2021年3月1日我院門診收入院。患者白天頻繁地抿嘴唇、做吞咽動作以及右側(cè)嘴角抽動,發(fā)作時意識清醒;偶有夜間熟睡中突然四肢屈曲伴抽動、大喊大叫、胡言亂語,持續(xù)十余秒后自行緩解,被家人喚醒,醒后無回憶,并否認做夢。追溯病史:10余年前因家人意外去世,有過2年的幻覺,表現(xiàn)為感覺已去世的親人站在背后;近十年來注意力、記憶力、計算力及語言理解能力逐漸變差,說話邏輯稍混亂、重復語言多,并有細微顫音,不能勝任工作及復雜的家務。就診時精神尚可,近期2 m內(nèi)體重減少20斤。既往史:2020年6月行卵巢囊腫手術(shù)。無特殊藥品服藥史。入院查體:神清,重復語言、有細微顫音,抿嘴、右側(cè)嘴角抽動,左手大拇指及食指末端細微的蚯蚓樣手足徐動樣不自主運動,雙上肢肌張力低,四肢肌力5級,雙側(cè)霍夫曼征及羅索里莫征陽性。家族史:該家系先證者的父親、叔叔、三個哥哥、侄子及堂姐均有類似癥狀,爺爺奶奶情況不詳,姥爺姥姥自然死亡,母親健在。父親及二哥癥狀較重,表現(xiàn)為典型舞蹈樣不自主運動,伴有精神行為異常,父親40歲時發(fā)病,50歲時去世(死因不詳),二哥43歲發(fā)病。先證者大伯、大哥、三哥及堂姐癥狀與其類似,以口咽部不自主運動為主,伴輕度認知功能下降,大哥49歲發(fā)病,三哥44歲發(fā)病,堂姐25歲發(fā)病(現(xiàn)已去世,死因不詳)。先證者侄子(三哥的兒子)14歲出現(xiàn)智能損害,隨之出現(xiàn)輕度的運動癥狀。患者與其丈夫(正常)育有1子1女,目前兒子初中,女兒高中,均體健。該家系圖譜如下(見圖1)。
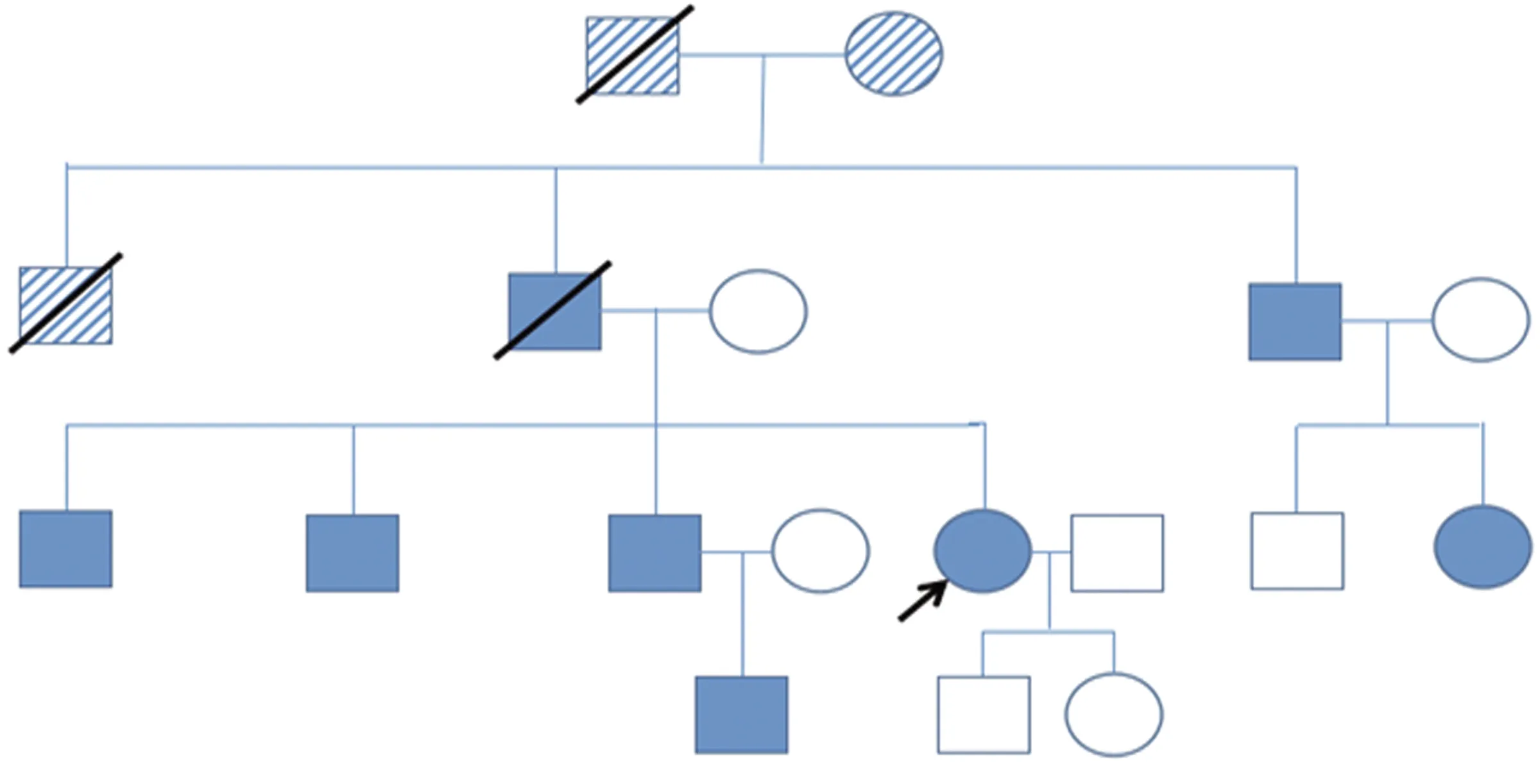
圖1 先證者家系圖譜
1.2 方法 (1)神經(jīng)電生理:采用40導聯(lián)數(shù)字化腦電圖儀(澳大利亞康迪Grael EEG),按國際10-20系統(tǒng)導聯(lián)安放法則記錄,設定敏感度100 μV/cm,低頻濾波0.3 Hz,高頻濾波20 Hz;(2)核磁共振:采用西門子1.5T核磁共振機對患者進行腦部平掃,掃描序列包括T1WI、T2WI、FLAIR、DWI及海馬磁共振成像;(3)認知功能檢測:簡易智力狀態(tài)檢查量表(MMSE)、Hamilton抑郁量表(HAMD)、蒙特利爾認知評估量表(MoCa)、臨床癡呆評定量表(CDR);(4)基因檢測:在先證者及家屬知情同意情況下,對先證者進行基因檢測。采取靜脈血4 ml,提取基因組DNA,由杭州漠柏醫(yī)學檢驗所進行HTT基因檢測。
2 結(jié) 果
2.1 腦核磁共振 腦萎縮,雙側(cè)海馬體積縮小,F(xiàn)LAIR序列信號增高,提示海馬硬化可能,左側(cè)海馬區(qū)脈絡膜裂囊腫(見圖2)。
2.2 神經(jīng)電生理 背景持續(xù)β節(jié)律。結(jié)論輕度異常成人腦電圖。
2.3 認知功能評定 MMSE:17分。MoCa:14分。Hamilton抑郁量表:7分。臨床癡呆評定量表CDR:1分,診斷:癡呆(輕度)。
2.4 裂隙燈下K-F環(huán) 陰性。
2.5 化驗 銅藍蛋白、腫瘤標志物、甲功5項、感染4項、生化及血、尿常規(guī)均正常。
2.6 基因檢測結(jié)果 檢測到IT15基因變異。先證者IT15基因的一個等位基因CAG重復次數(shù)為16次,屬正常范圍;另一個CAG重復次數(shù)為44次,在全突變范圍內(nèi)(見圖3)
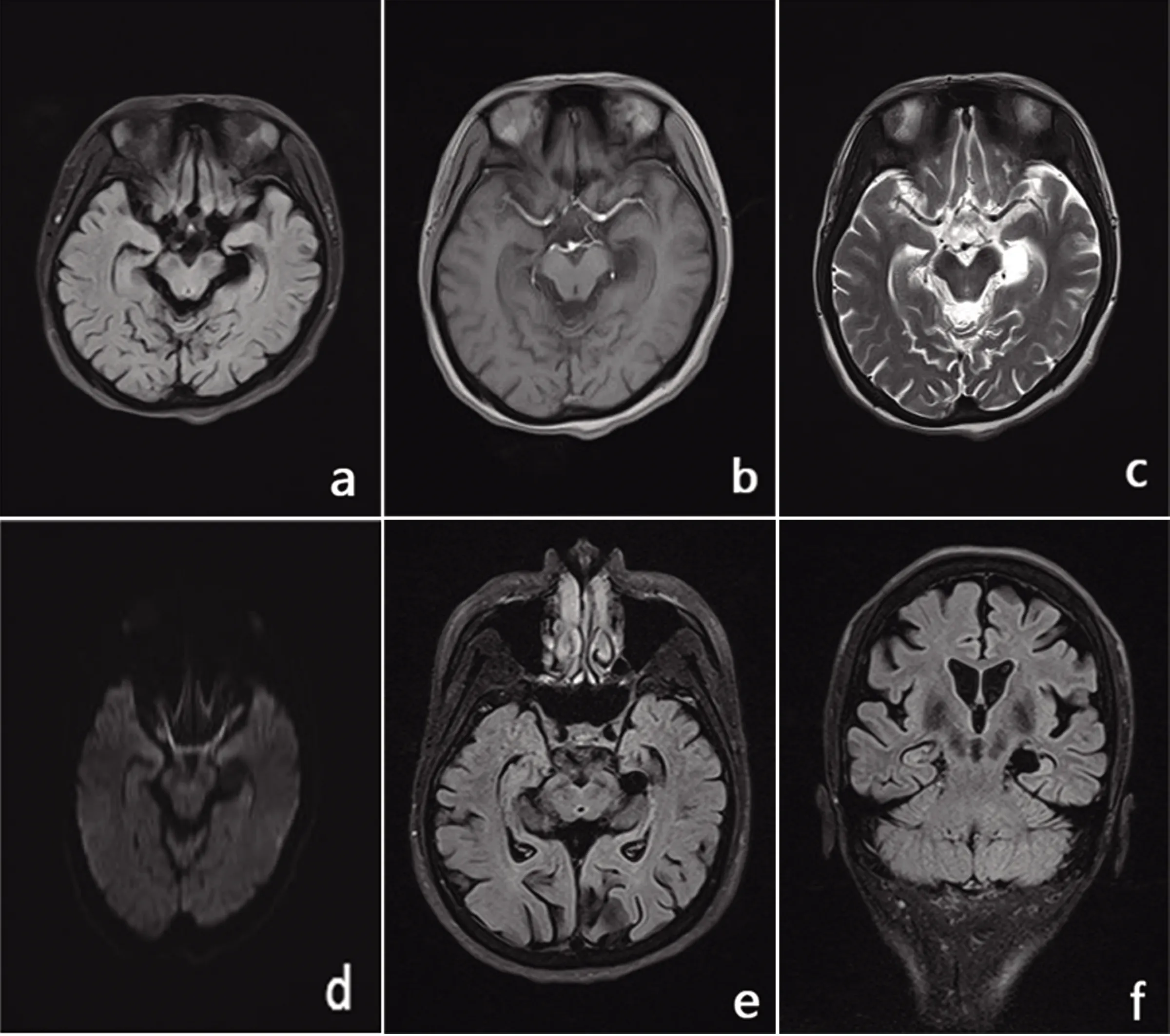
圖2 a:FLAIR;b:T1;c:T2;d:DWI;e、f:海馬磁共振成像:腦溝池裂增寬,雙側(cè)海馬體積縮小,F(xiàn)LAIR序列信號增高,左側(cè)海馬區(qū)可見長T1長T2信號影,邊界清晰
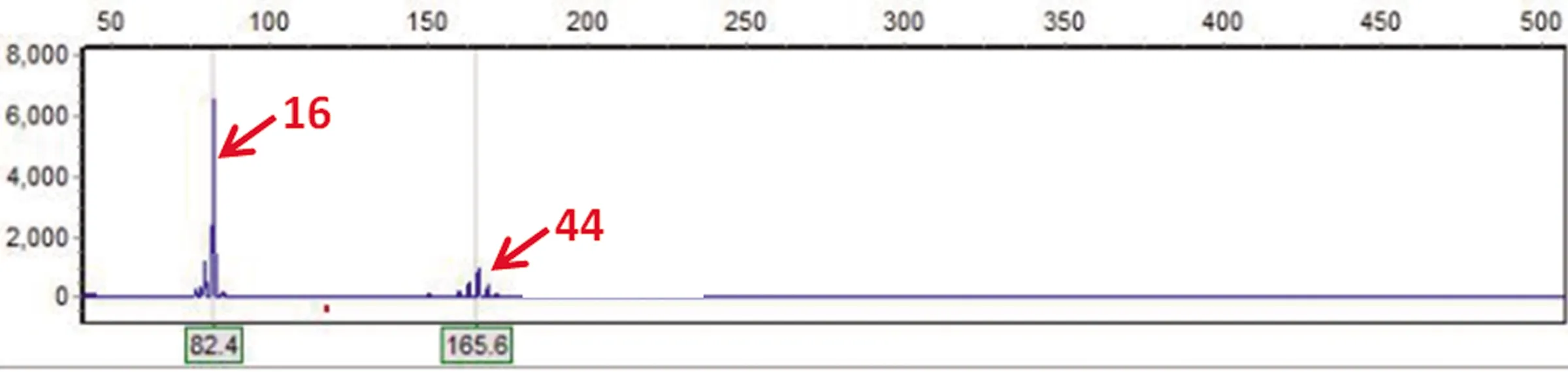
圖3 先證者HTT基因檢測CAG重復數(shù),一個為16次,屬于正常范圍;另一個為44次,屬于全突變范圍
3 討 論
Huntingtong病北美、歐洲和澳大利亞的患病率為5.70/10萬人,在亞洲為0.40/10萬人[1]。HD是第4號染色體上的IT15基因突變引起N端編碼區(qū)內(nèi)的三核苷酸(CAG)重復序列拷貝數(shù)異常增多,產(chǎn)生突變型亨廷頓蛋白(mHtt)導致的遺傳變性病[2]該病典型臨床表現(xiàn)主要涉及運動障礙、精神異常及認知障礙三大方面。Huntingtong病理改變以基底節(jié)區(qū)的尾狀核、殼核以及大腦皮質(zhì) (特別是額葉) 萎縮為主;神經(jīng)細胞脫失亦可累及丘腦腹外側(cè)核、下丘腦、黑質(zhì)網(wǎng)狀結(jié)構(gòu)、橄欖體、薄束核和楔束核、白質(zhì)和間腦核等部位。Masnata等[3]的研究顯示,tau病理證據(jù)支持將HD歸類為繼發(fā)性tau病,tau功能障礙是否直接導致一些與HD相關(guān)的特征,特別是認知缺陷尚需深入研究,有研究者認為tau蛋白和突變的Huntingtin蛋白(mHTT)共同作用于神經(jīng)退行性變和認知功能障礙。
該家系共有HD患者8人,其中先證者父親及二哥2人運動癥狀最重,表現(xiàn)為典型舞蹈樣不自主運動,伴有明顯精神行為異常,父親40歲時發(fā)病,50歲時去世(死因不詳),二哥43歲發(fā)病;先證者及其大伯、大哥、三哥、堂姐共5人的運動癥狀較輕,以口咽部不自主運動為主,伴輕度認知功能下降,先證者同時存在左手大拇指及食指末端細微的蚯蚓樣手足徐動樣不自主運動,大伯45歲發(fā)病,大哥49歲發(fā)病,三哥44歲發(fā)病,堂姐25歲發(fā)病(現(xiàn)已去世,死因不詳);先證者侄子14歲發(fā)病,以智能障礙為首發(fā)癥狀,隨后出現(xiàn)與先證者相似的運動癥狀。
該家系先證者在出現(xiàn)運動癥狀前多年即出現(xiàn)了精神及認知方面的異常,患者的侄子14歲時首先出現(xiàn)智能的減退。一系列研究[4~13]的結(jié)論可以很好解釋該現(xiàn)象:HD患者的非運動癥狀例如觀念運動性失用、思維變緩及執(zhí)行功能障礙等,可以在就診前多年即出現(xiàn)。這與CAG重復數(shù)相關(guān),首先表現(xiàn)為認知障礙的HD患者年齡較小、CAG重復數(shù)目較多,青少年和成人HD患者運動癥狀發(fā)病年齡(AAO)與CAG重復數(shù)呈負相關(guān),老年HD患者的CAG重復數(shù)與AAO無相關(guān)性。這與HD的基因動態(tài)突變及遺傳早現(xiàn)有關(guān)。
該家系中患者運動癥狀的多樣性與既往文獻報道[14,15]相一致,舞蹈樣癥狀為其典型表現(xiàn),還可見肌張力障礙(斜頸、角弓反張、弓足等)、姿勢反射消失、運動遲緩和肌強直等非舞蹈樣癥狀,但相關(guān)報道較少。在HD患者因明顯運動癥狀就診前數(shù)年,即可出現(xiàn)較細微、不易察覺的臨床前運動表現(xiàn),包括眼球運動、步態(tài)和精細運動表現(xiàn)的變化等,這種細微的、非特異性的運動癥狀常被忽略,易漏診或誤診。雷爾曼等于2014年依據(jù)患者臨床特征提出HD可分為癥狀前期、前驅(qū)期和癥狀期,癥狀期又可分為早期、中期和晚期。癥狀前期,致病基因攜帶者沒有任何運動、認知及精神方面的癥狀。前驅(qū)期,致病基因攜帶者多表現(xiàn)為性格及情緒變化等精神癥狀。癥狀期早期可以存在輕微的運動、精神或認知癥狀,隨著疾病進展,患者的運動癥狀逐漸明顯。
HD的病理改變主要為大腦皮質(zhì)及雙側(cè)基底節(jié)區(qū)萎縮,于是CT/MRI上表現(xiàn)為大腦皮質(zhì)和尾狀核萎縮,腦室擴大。該家系先證者頭部MRI提示腦溝裂池增寬,腦萎縮,符合HD影像學改變。據(jù)此有研究[16]認為CT/MRI上表現(xiàn)的 “蝴蝶征”屬于HD特征性的改變,但特異性較低。HD影像學改變還包括:(1)頭部PET/CT掃描可見雙側(cè)基底節(jié)、大腦各葉皮質(zhì)等部位代謝減低,而且先于尾狀核萎縮形態(tài)學改變,有利于 HD的早期診斷。(2)MR波譜基底節(jié)區(qū)NAA/Cr(N-乙酰門冬氨酸/肌酸)值明顯減低,Cho/Cr(膽堿/肌酸)值明顯增高,異常乳酸 (Lac)峰。HD患者腦電圖呈彌漫性異常,無特異性。
HD往往根據(jù)發(fā)病年齡,臨床癥狀及家族史可以作出臨床診斷,對于無家族史或者臨床表現(xiàn)不典型的HD患者,通過檢測IT-15基因CAG重復次數(shù)可以確診。目前基因診斷標準(美國醫(yī)學遺傳學會(ACMG)制定的HD基因測試技術(shù)標準語指南(2004版)如下:正常基因的CAG重復次數(shù)≦26;當CAG重復次數(shù)為27∽35時,尚不足以引起臨床癥狀,但基因不穩(wěn)定,在通過精子傳遞給下一代時,可出現(xiàn)CAG重復次數(shù)的擴增;當CAG重復次數(shù)為36∽39時,具備不完全外顯率,部分攜帶者可不發(fā)病或推遲發(fā)病時間;當CAG重復次數(shù)≧40時,具備完全外顯率,所有攜帶者均發(fā)病。HD基因測試陽性定義為至少1個等位基因的CAG重復次數(shù)≧40,具有99%以上的敏感度和100%的特異度。該家系先證者臨床表現(xiàn)不典型,行HTT基因檢測,結(jié)果顯示:HTT一個等位基因CAG重復數(shù)為44次,屬于全突變范圍,確診HD。
予以先證者硫必利片(50 mg,tid)、丁苯酞軟膠囊(200 mg,tid)及B族維生素對癥治療后,患者動癥狀較入院時改善。現(xiàn)階段HD的治療仍以對癥治療為主,能夠緩解患者癥狀,但無法延緩病程進展。近年來HD的分子遺傳學治療引起了國內(nèi)外學者的廣泛關(guān)注,雖然尚無法實現(xiàn),但為HD的治療指明了新的方向。
由于在亨廷頓病(HD)的疾病過程中,代謝的改變早于形態(tài)學改變,而隨著紋狀體的病變達到一定程度之后逐漸出現(xiàn)臨床運動癥狀,因此,當出現(xiàn)了HD的臨床表現(xiàn),特別是臨床前運動癥狀或精神、認知障礙,或者有明確家族史,應本著早發(fā)現(xiàn)、早診斷、早治療的原則,及早就醫(yī),采取恰當?shù)纳窠?jīng)保護措施以防止主要神經(jīng)元的丟失,延緩疾病進程、提高生活質(zhì)量。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫(yī)苑(2022年1期)2022-08-30 08:39:40
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
家庭醫(yī)學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫(yī)藥(2020年10期)2020-02-13 15:45:52
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
