Cl-在α-Al2O3表面吸附的馳豫方式和溶劑化模型選擇
2022-03-04 13:23:06高新宇祖武杰
原子與分子物理學報 2022年3期
高新宇, 祖武杰, 伍 斌, 董 瀚,2,史 文, 王 洋
(1. 上海大學 材料科學與工程學院, 上海 200444; 2. 鋼鐵研究總院, 北京 100081; 3. 上海大學 計算機工程與科學學院, 上海 200444)
1 引 言
鋁合金由于被動氧化膜的保護在大部分環(huán)境中表現(xiàn)出了良好的耐腐蝕性能. 然而,部分環(huán)境中的氯離子(Cl-)等鹵素陰離子會引發(fā)氧化鋁膜(Al2O3)的局部降解,進而導致合金表面的點蝕,造成材料的破壞失效[1-4]. 關于Cl-與Al2O3之間相互作用的機制,目前主要有三種觀點[4-7]:1)Cl-遷移/滲透到金屬與氧化物界面導致局部破壞[7];2)Cl-和O2-的競爭性吸附[8,9];3)氧化物的破壞與修復[10]. 這些觀點普遍認為Cl-與氧化膜的相互作用是點蝕形成的起始原因. 氯化物滲透到氧化膜的第一步是對表面離子的吸附,然后是進一步的遷移或反應[4,6,9,11].
用計算的方法研究Cl-在Al2O3表面上吸附的報道,目前已經(jīng)有了一些. 例如,Liu等[11]和Zhang等[12]計算了Cl-在α-Al2O3(0001)和Al(111)表面內的遷移和Cl-在α-Al2O3上的吸附. Zhang等[13,14]研究了H2O和Cl-對層缺陷氧化鋁的侵蝕,以及OH、Cl和H2O在層缺陷Al2O3膜上的共吸附行為. Marks等[15]計算分析了水溶液中的Cl-對羥基化α-Al2O3(001)和(100)表面的腐蝕. 這些報道做了細致的工作,從微觀角度解釋了相關實驗現(xiàn)象. 然而,作為被研究對象,α-Al2O3(0001)表面模型馳豫方式的選擇和水溶液模型的選用可能對計算結果造成的影響,沒有被詳細地評估.
離子在表面上的吸附過程會造成表面多層原子的弛豫,這可能導致部分馳豫模型(部分原子被固定的模型)的計算結果與實際產(chǎn)生偏差. 特別是對于α-Al2O3等離子性強的金屬氧化物,長程靜電效應的中斷和人工偶極矩的產(chǎn)生可能會導致部分弛豫結構和完全弛豫結構之間的顯著差異,最終造成計算結果的大幅偏差[16-18]. Al、Pt等陽離子在α-Al2O3表面的吸附計算結果表明,不同的弛豫模型會導致吸附結構和離子吸附能[16]的差異. 然而,模型馳豫方式對Cl-與Al2O3之間相互作用的影響卻鮮有報道. 因此,有必要探討Cl-在α-Al2O3表面吸附計算中的模型選擇.
在鋁合金等金屬表面的點蝕過程中,水溶液對表面的化學/電化學反應有重要的影響. 因此,需要一種有效且準確的方法來模擬材料表面水環(huán)境. 在密度泛函理論(DFT)計算中,主要有顯式溶劑化模型和隱式溶劑化模型兩種處理溶液的方法. 其中隱式溶劑化模型可以較好地模擬溶劑的長程靜電相互作用和平均性質,在計算和模型改進中具有良好的實用性[19].
本工作采用第一性原理的方法,計算Cl-在α-Al2O3(0001)表面的吸附行為. 通過設置不同的弛豫模型,研究弛豫方式對氧化鋁表面性質和Cl-在表面吸附行為的影響. 計算考慮了真空環(huán)境和水溶液環(huán)境,將得出的計算結果與實驗數(shù)據(jù)對比,評估了溶劑化模型的作用. 對于Cl-與α-Al2O3(0001)相互作用的更多研究,本文在前期的模型馳豫方式選擇和溶劑化模型設置方面提供了一種可靠的思路.
2 計算方法與模型
2.1 計算方法
計算在基于密度泛函理論(Density functional theory, DFT)的VASP(Vienna ab-initio simulation package)[20]上運行. 交換關聯(lián)泛函的描述基于廣義梯度近似(Generalized gradient approximation, GGA)[21]的Perdew-Burke-Ernzerhof(PBE)[22],離子-電子的相互作用采用PAW(Projector augmented wave)描述[23]. 布里淵區(qū)的k點用Monkhorst-Pack方法采樣[24].
α-Al2O3單胞的優(yōu)化以0.0001 eV為能量收斂標準,最終的平面波截止動能取650 eV,k點為15×5×5. 優(yōu)化后的晶格參數(shù)為a=b=4.808 ?,c=13.123 ?,與其它實驗[25]或計算[18]研究中的差值在1%以內. 在表面層的優(yōu)化計算中,原子層數(shù)和真空層厚度的選取以α-Al2O3(0001)表面能的變化小于0.01 J/m2為標準. 所有Cl-在表面的吸附計算中,平面波截斷能取 650 eV,k 點取 8×8×1. 電子自洽計算收斂的標準為10-4eV,當體系中每個原子的應力小于0.01 eV/?時,馳豫終止. 計算首先在真空條件下運行,然后在VASPsol方法[26]模擬的水溶液環(huán)境下進行,水的介電常數(shù)設置為標準值80[19]. 為了研究弛豫方式對計算結果的影響,我們使用了固定底部六層原子的部分弛豫結構和允許所有原子馳豫的完全弛豫結構.
Cl-在α-Al2O3(0001)表面的吸附能由下式計算:
Eads,Cl=ECl/Al2O3- (EAl2O3+ECl)
(1)
其中ECl/Al2O3表示Cl-吸附到表面后體系的總能量,EAl2O3是清潔表面(未吸附Cl-的表面)的能量,ECl表示孤立Cl原子的能量.
2.2 理論模型
本文使用12層原子和15 ?真空層的(2×2)α-Al2O3(0001)表面模型. 全弛豫模型(12-R)和部分馳豫模型(12-C)如圖1(a)、(b)所示. 在圖1(a)中,Al1表示第一層鋁原子,O2表示第二層氧原子,以此類推. 對于Cl-在表面的吸附,我們考慮了5個不同的位點,如圖1(c)所示. 位點名稱是以Cl-正下方的原子命名的,編號代表原子所在層數(shù),遵循Hernández[27]的命名法.

圖1 (a)完全弛豫和(b)部分弛豫的α-Al2O3(0001)表面模型側視圖;(c)Cl-吸附位點俯視圖.紅色原子表示O,灰色原子表示Al.Fig. 1 Side views of the (a) full relaxation and (b) partial relaxation α-Al2O3 (0001) models, and (c) top view of the adsorption sites of Cl- on the surface. The red and grey atoms are O and Al, respectively.
3 結果與討論
3.1 弛豫方式和溶劑化模型對α-Al2O3(0001)表面結構的影響
表1統(tǒng)計了12-R和12-C在真空和水溶液環(huán)境下優(yōu)化后的層間間距百分比變化,并列出了其它計算和實驗工作中的相應變化,其中“+”表示平均原子層間距的增加,“-”表示減少. 可以看出,兩種弛豫模型的原子層在真空和溶劑化條件下均有較大的弛豫量,其中d(Al1-O2)的變化值最大,第一層Al原子(Al1)向下移動到幾乎與第二層O原子(O2)共面,這和其它研究中的結果相近.

表1 真空和水溶液環(huán)境下12-R和12-C的原子層層間距變化
從馳豫方式的角度來看,真空和溶液條件下,優(yōu)化后的12-R和12-C模型原子層層間距發(fā)生了明顯變化. 其中真空下12-C的d(O2-Al3)比12-R增大了60%;溶液環(huán)境中,12-R和12-C模型的d(Al1-O2)差值超過13%,其它的層間距也有不同程度的改變. 這些變化說明12-C模型中被固定的底層6層原子對體系最終結構有較大影響,這可能會影響到后續(xù)計算結果的可靠性.
從溶劑化模型的角度來看,真空條件下兩種馳豫模型的原子層層間距值與實驗值有很大差距. 而在水溶液條件下,12-R和12-C中d(Al1-O2)和d(O2-Al3)的變化量分別為-66.0%、-79.0%和+7.7%、+9.3%,更接近實驗值的-51%、+16%. 對于溶液條件下兩種結構的d(Al3-Al4)和d(Al4-O5),雖然和實驗值有一定差異,但是和真空條件下的結果沒有顯著區(qū)別,并且第4、5層原子對Cl-在外表面的吸附影響較小. 以上結果表明,在VASPsol隱式溶劑化模型下優(yōu)化得到的α-Al2O3(0001)表面更接近實驗結果,是較為可靠的理論模型.
3.2 弛豫方式和溶劑化模型對Cl-在α-Al2O3(0001)表面吸附行為的影響
表2統(tǒng)計了Cl-在真空和水溶液環(huán)境下的12-R和12-C表面不同吸附位點上的吸附能、吸附距離和吸附方式. 當吸附狀態(tài)為化學吸附(cp)時,吸附距離為Cl-與被吸附原子之間的鍵長;當吸附狀態(tài)為物理吸附(pp)時,統(tǒng)計的吸附距離為Cl-與第一層原子之間的平均距離.
在真空和水溶液條件下,Cl-在12-R和12-C模型上均有三種穩(wěn)定吸附結構和兩種吸附狀態(tài).真空條件下Cl-在12-R和12-C表面最穩(wěn)定的吸附位點均為Al1(吸附結構如圖2(a)和(c)所示),吸附距離為2.14 ?,吸附能分別為-2.20 eV和-2.27 eV. 在溶液環(huán)境下,Al1位點仍然是12-R和12-C表面最穩(wěn)定的吸附位點(吸附結構如圖2(b)和(d)所示),其吸附能分別為-3.05 eV和-3.16 eV,吸附距離分別為2.30 ?和2.31 ?. Cl-在所有Al3和Al4位點均為物理吸附. 在能量最小化過程中,O2和O5位上的Cl-是不穩(wěn)定的,會自發(fā)地向Al1位移動,最終產(chǎn)生和Al1位點同樣的吸附結構,這與其它計算的結果相同[12].
綜合對比真空和水溶液條件下的結果可知,水環(huán)境中Cl-在所有位點的吸附距離均有一定幅度增加,吸附能有明顯下降. Cl-在VASPsol方法模擬的水溶液環(huán)境中有更低的吸附能,這說明Cl-在液相環(huán)境中有更強的吸附驅動力,更容易與氧化鋁表面之間發(fā)生相互作用,這與常見的腐蝕現(xiàn)象相符合. 此外,對于12-R和12-C模型,Cl-在真空或溶液環(huán)境中相同位點的吸附能和吸附距離均有不同幅度的區(qū)別,其中水溶液中Cl-在兩種馳豫模型Al3位點上的吸附能和吸附距離相差達到30%和54%. 由此可見,在Cl-吸附到表面的計算中,采用部分馳豫的模型仍然會對結果產(chǎn)生較為明顯的影響.
為了進一步了解Cl-與氧化鋁外表面的相互作用,并研究模型馳豫方式對系統(tǒng)電荷分布的影響. 部分結構的電子局域函數(shù)(Electron Localization Function, ELF)和差分電荷圖被展示在圖3和圖4中.

圖3 水溶液條件下,12-R和12-C三種吸附結構的ELF:(a)12-R-Al1;(b)12-R-Al3;(c)12-R-Al4;(d)12-C-Al1;(e)12-C-Al3;(f)12-C-Al4.Fig. 3 ELFs of three adsorption structures of full and partial relaxations in solvation environment, (a) 12-R-Al1; (b) 12-R-Al3; (c) 12-R-Al4; (d) 12-C-Al1; (e) 12-C-Al3; (f) 12-C-Al4.
ELF被廣泛用于描述分子中的化學鍵,其標準化數(shù)值在0到1之間.取上限值1表示電子完全局域化,0.5表示電子完全離域化[30,31].圖3(a)和(d)分別是溶液條件下12-R和12-C結構Al1位點吸附Cl-的ELF,ELF值為0.856,可以看出Cl-與Al之間存在電子高度局域化區(qū)域,且其ELF值大于0.85. 圖4(a)和(d)的差分電荷圖也表明,在兩種結構中,Cl-與氧化鋁表面原子之間均存在明顯的電荷交換. 這些結果表明12-R和12-C的相應結構中,Al與Cl-之間存在較強的共價鍵作用. 表3中統(tǒng)計了水溶液環(huán)境下12-R和12-C的清潔表面和Cl-吸附表面第一層四個Al和第二層三個O的bader電荷,Al和O原子的位置及名稱如圖5所示. 從表3中可以看出,兩種馳豫結構中被吸附的Al原子(Al1(1))的bader電荷均有所增加,而第一層其它三個Al和Al1(1)附近的三個O的bader電荷則有所減小. 這一變化證明了Cl-吸附到表面Al1(1)上之后,第一層其它Al及附近O原子的電荷均向“Al-Cl”單元轉移,Cl-與表面之間發(fā)生了較強的相互作用.
圖3(b)、(c)、(e)、(f)中的ELF表明,在12-R和12-C的Al3和Al4位點上,Cl-和α-Al2O3(0001)表面的任何原子之間均沒有電子局域化區(qū)域. 圖4(b)、(c)、(e)、(f)的差分電荷圖也表明Cl-和表面之間沒有明顯的電荷交換現(xiàn)象. 但是對比清潔表面,Cl-在這些位點的吸附實際上導致了第一層Al和第二層O的bader電荷減少. 這表明Cl-與表面之間存在較弱的相互作用,Cl-在表面發(fā)生了物理吸附. 值得注意的是,雖然12-R和12-C對照結構中同一原子的bader電荷值均有一定的差異,但是沒有影響到對Cl-與表面相互作用分析的整體結果.
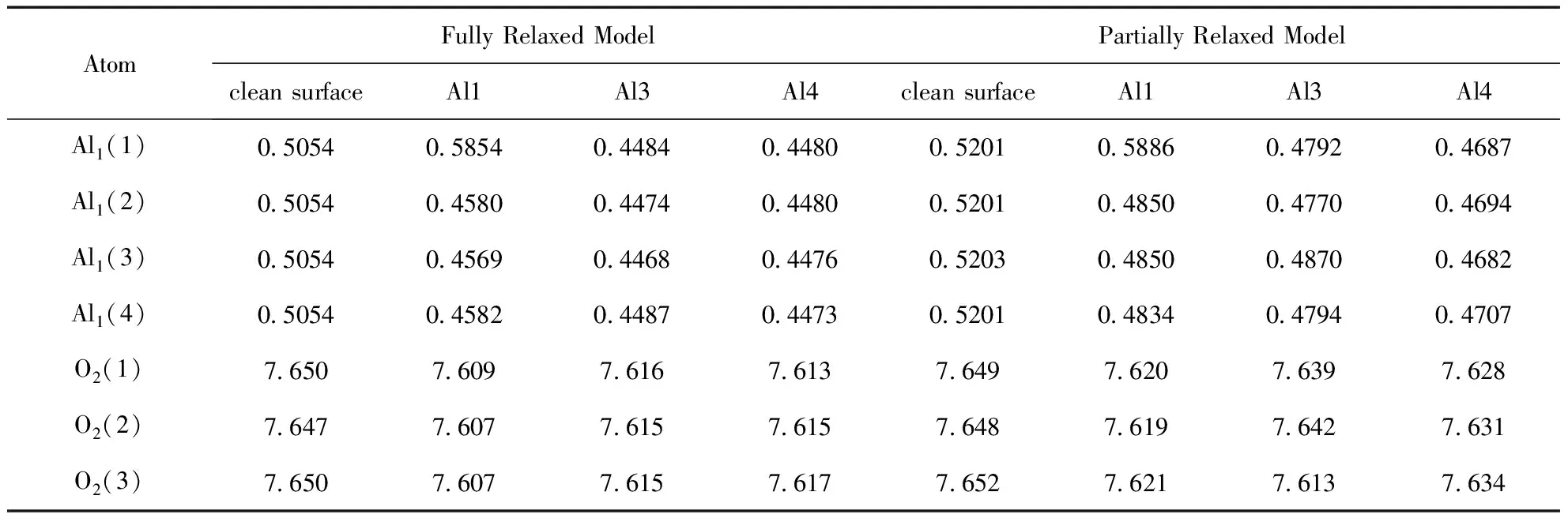
表3 水溶液條件下,12-R和12-C的最穩(wěn)定吸附結構中第一層Al和第二層O的bader電荷

圖5 表3中使用的原子代號示意圖Fig. 5 Schematic diagram of atomic markers used in Table 3.
4 結 論
本文通過設置12-R和12-C兩種模型馳豫方式,在真空和VASPsol方法模擬的水溶液條件下,計算分析了α-Al2O3(0001)的表面結構和Cl-在表面的吸附行為,得出了以下結論.
(1)固定底層6層原子的方式在真空和水溶液條件下均導致了α-Al2O3(0001)表面結構的畸變. 在后續(xù)的吸附計算中,雖然其沒有對Cl-在表面的吸附方式和體系電荷變化的趨勢造成影響,但是造成了具體的吸附能、吸附距離和bader電荷值差異. 在氧化鋁表面的點蝕研究中,這可能會造成后續(xù)更多的計算結果偏差. 因此,本文建議使用全弛豫的表面模型.
(2)在水溶液條件下,α-Al2O3(0001)的表面結構更接近于實驗測量值,且Cl-在表面有更強的吸附驅動力,這和常見的腐蝕現(xiàn)象相符合. 采用隱式溶劑化模型中的VASPsol方法,并設置一個合理的介電常數(shù),是較為可靠的模擬材料表面水溶液環(huán)境的方法.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中老年保健(2021年12期)2021-08-24 03:30:40
哲學評論(2021年2期)2021-08-22 01:53:34
中國傳媒大學學報(自然科學版)(2021年1期)2021-06-09 08:43:00
中學生數(shù)理化·七年級數(shù)學人教版(2020年10期)2020-11-26 08:24:50
數(shù)學物理學報(2020年2期)2020-06-02 11:29:24
中國生殖健康(2020年6期)2020-02-01 06:28:50
中華詩詞(2019年7期)2019-11-25 01:43:04
中國生殖健康(2019年11期)2019-01-07 01:28:02
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
