HPLC法測定水合羥基化頭孢克洛雜質研究
2022-02-19 08:46:32劉萍胡利敏田洪年李敏張文勝楊夢德賈全孫玉雙
中國抗生素雜志 2022年1期
劉萍 胡利敏 田洪年 李敏 張文勝 楊夢德 賈全 孫玉雙
(華北制藥河北華民藥業有限責任公司,石家莊 052165)
頭孢克洛(cefaclor)是第二代口服頭抱菌素衍生物,是EliLilly公司在1975年報道的新藥[1],是廣泛應用于抗感染的頭孢菌素類抗生素藥物。頭孢克洛抗菌譜主要是:葡萄球菌、肺炎雙球菌、大腸埃希菌等,其作用機制為使轉肽酶失活,干擾細菌細胞壁的合成,阻止黏肽原的交聯[2]。頭孢克洛雜質包括合成過程中產生的工藝雜質以及在酸、堿、高溫和光照條件下產生的降解雜質[3-4]。頭孢克洛在高溫和強光條件下均會降解產生羥基化降解物雜質。頭孢克洛和水合羥基化頭孢克洛雜質結構式見圖1。水合羥基化頭孢克洛雜質的化學名為(6R, 7R)-7-(R)-2-氨基-2-苯乙酰氨基)-3-氯-2-羥基-8-氧代-5-硫雜-1-氮雜雙環[4.2.0]辛-3-烯-2-羧酸。水合羥基化頭孢克洛雜質未被《歐洲藥典》10.0版(EP10.0)[5]收載在頭孢克洛原料藥項下。《中國藥典》2020版收載了頭孢克洛原料藥的有關物質測定方法,未對各雜質進行單獨控制,未對水合羥基化頭孢克洛雜質準確定性定量研究,參照《中國藥典》2020版頭孢克洛原料藥等[6-7]有關物質方法對該雜質進行測定,并對該方法進行方法學考察,同時采用加校正因子的主成分自身對照法和外標法對該雜質進行定量研究。試驗證明該方法操作簡便、準確度高、重現性好,可以采用不加校正因子的主成分自身對照法進行定量。
1 儀器與試藥
1.1 儀器
1260型高效液相色譜儀(DAD檢測器、美國Agilent公司);XS105電子分析天平(瑞士Mettler-Toledo公司);FE20 pH計(瑞士Mettler-Toledo公司)。
1.2 試藥
頭孢克洛對照品(中國食品藥品檢定研究院,批號130418-201606,含量94.4%);水合羥基化頭孢克洛雜質對照品(廣州牌牌生物科技有限公司,批號PITBKL-2-20190112-03,含量95.17%);頭孢克洛原料藥(華北制藥河北華民藥業有限責任公司,批號D1121903002);磷酸二氫鈉、磷酸為分析純,乙腈為色譜純,水為純化水。
2 色譜條件
2.1 檢測波長的選擇
取水合羥基化頭孢克洛雜質對照品用0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋制成0.1 mg/mL的溶液,在190~400 nm范圍紫外掃描,見圖2。譜圖顯示,水合羥基化頭孢克洛雜質在220 nm處有最大吸收,與《中國藥典》2020版收載的頭孢克洛原料藥的有關物質測定方法中檢測波長一致。
2.2 色譜條件
參照《中國藥典》2020版收載的頭孢克洛原料藥的有關物質測定方法。色譜柱:資生堂CAPCELL PAK AQ C18(4.6 mm×250 mm,5 μm);流動相A為0.78%磷酸二氫鈉溶液(取磷酸二氫鈉7.8 g,加水溶解并稀釋至1000 mL,用磷酸調pH至4.0),流動相B為0.78%磷酸二氫鈉溶液(pH4.0)-乙腈(55:45);流速1.2 mL/min;檢測波長:220 nm;柱溫:25℃;進樣量20 μL;按表1進行線性梯度洗脫。

表1 梯度洗脫時間表Tab.1 Gradient elution schedule
2.3 溶液配制
2.3.1 對照品溶液
精密稱取水合羥基化頭孢克洛雜質對照品6 mg,置于50 mL量瓶,加0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋至刻度,搖勻。
2.3.2 供試品溶液
取頭孢克原料藥約50 mg,精密稱定,置10 mL量瓶中,加0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋至刻度,搖勻。
2.3.3 系統適用性溶液
取供試品約50 mg,精密稱定,置10 mL量瓶中,精密量取對照品溶液1.5 mL置上述量瓶中溶解再加0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋至刻度,搖勻。
2.4 系統適用性試驗
精密量取系統適用性溶液20 μL,進樣檢測,記錄色譜圖。結果見圖3,頭孢克洛與水合羥基化頭孢克洛雜質分離度為4.67,大于1.5,證明該方法系統適用性良好。
3 校正因子的測定
3.1 雜質峰定位
取“2.3.3”項下系統適用性溶液,按“2.2”項下色譜條件進行測定,記錄色譜圖,確定水合羥基化頭孢克洛雜質的保留時間,計算該雜質相對于主成分頭孢克洛的相對保留時間(RRT=t雜質/t頭孢克洛),結果為1.11,見圖3。
3.2 雜質校正因子的測定
3.2.1 重復性試驗
精密稱取頭孢克洛對照品和水合羥基化頭孢克洛雜質對照品適量,用0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋制得濃度分別為定量限、0.010、0.020、0.025、0.030和0.035 mg/mL的系列標準溶液,分別進樣,按“2.2”項下色譜條件進行測定,記錄色譜圖。以對照品溶液濃度C為橫坐標,色譜峰面積A為縱坐標,進行線性回歸。
分別配制3組頭孢克洛、水合羥基化頭孢克洛雜質線性濃度,繪制標準曲線,計算線性回歸方程,頭孢克洛與雜質回歸方程式斜率之比即為校正因子,并計算校正因子的RSD值。
頭孢克洛和水合羥基化頭孢克洛雜質線性重復性結果見表2,水合羥基化頭孢克洛雜質校正因子計算重復性結果見表3。

表2 線性重復性結果Tab.2 Linear repeatable result

表3 校正因子計算重復性結果Tab.3 Correction factor calculation repeatability results
3.2.2 中間精密度試驗
不同人員在不同日期使用不同的儀器,分別配制3組頭孢克洛、水合羥基化頭孢克洛雜質線性曲線,通過計算頭孢克洛與水合羥基化頭孢克洛雜質回歸方程式斜率之比,求得水合羥基化頭孢克洛雜質的校正因子,并計算校正因子的RSD值。具體試驗過程同“3.2.1”項。
頭孢克洛和水合羥基化頭孢克洛雜質線性中間精密度結果見表4,水合羥基化頭孢克洛雜質校正因子計算中間精密度結果見表5。

表4 線性中間精密度結果Tab.4 Linear intermediate precision results
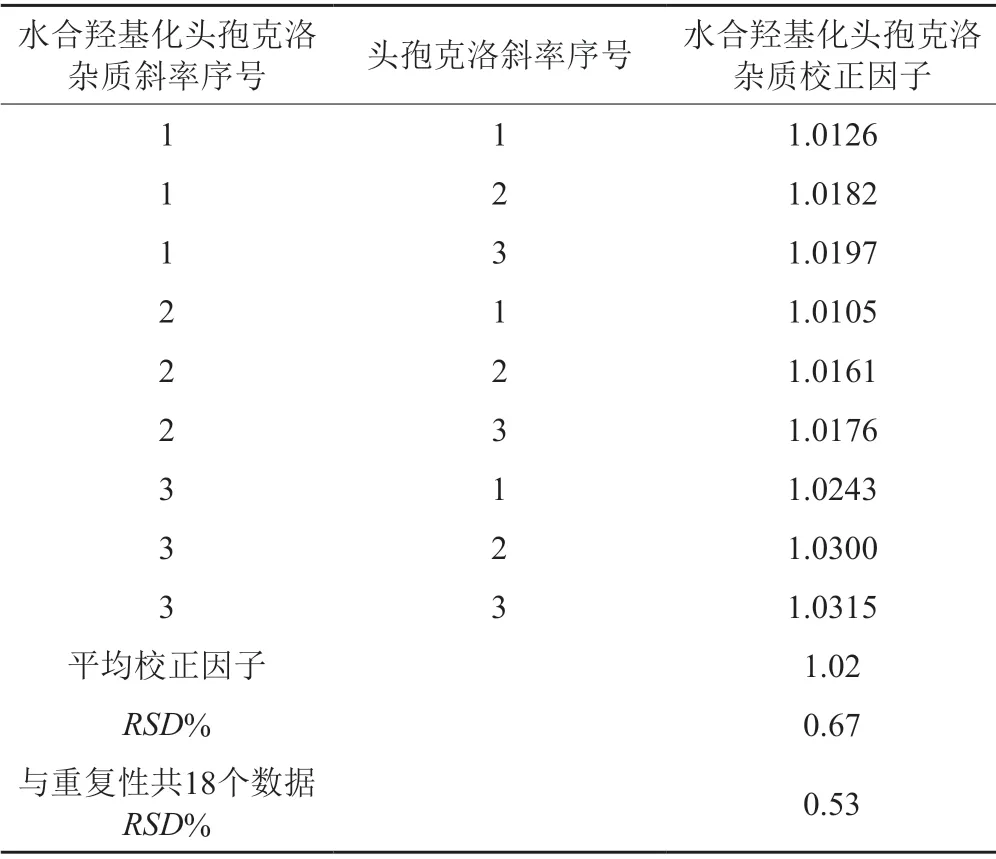
表5 校正因子計算中間精密度結果Tab.5 Correction factor to calculate intermediate precision results
結果表明:重復性和中間精密度中9個校正因子的RSD均小于5.0%;中間精密度與重復性數據比較,計算出水合羥基化頭孢克洛雜質校正因子的18個數的RSD為0.53%,小于5.0%,說明該方法測定水合羥基化頭孢克洛雜質校正因子中間精密度良好。水合羥基化頭孢克洛雜質校正因子為1.02。
4 方法學考察
4.1 專屬性試驗
以0.27%磷酸二氫鈉溶液(pH2.5)為溶劑,按“2.2”項下色譜條件進樣檢測,記錄色譜圖。在頭孢克洛和水合羥基化頭孢克洛雜質處均無色譜峰出現,表明溶劑無干擾(圖略)。
4.2 檢測限和定量限
精密量取對照品溶液逐級稀釋,按“2.2”項下色譜條件進樣測定,結果水合羥基化頭孢克洛雜質的檢測限(S/N=3.5)為5.5594ng,相當于供試品溶液濃度(5 mg/mL)的0.05559‰;定量限(S/N=10.5)為17.721ng,相當于供試品溶液濃度(5 mg/mL)的0.1772‰。取定量限溶液重復進樣6次,主峰峰面積的RSD為3.47%,主峰保留時間的RSD為0.16%。
4.3 線性試驗
具體試驗過程見“3.2.1”項。水合羥基化頭孢克洛雜質的回歸方程為A=22012.3009c-0.9319。結果表明,水合羥基化頭孢克洛雜質在9.108×10-4~3.400×10-2mg/mL范圍內線性關系良好(r=1.0000)。
4.4 回收率試驗
4.4.1 試驗過程
精密量取“2.3.1”項下對照品溶液5 mL,置25 mL量瓶中,用0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋至刻度,搖勻,作為外標法對照品溶液。
按“2.3.2”項下方法制備供試品溶液。
精密量取“2.3.1”項下供試品溶液1 mL,置100 mL量瓶中,用0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋至刻度,搖勻,作為1%對照品溶液。
精密稱取頭孢克洛原料藥(批號D1120903002,水合羥基化頭孢克洛雜質含量為0)約50 mg,共9份,分別置10 mL量瓶中,平均分成3組,每組分別加入“2.3.1”項下對照品溶液1.6、2.0和2.4 mL,用0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋至刻度,搖勻,作為回收率溶液。
分別精密量取外標法對照品溶液、供試品溶液、1%對照品溶液和回收率溶液各20 μL注入液相色譜儀,按“2.2”項下色譜條件進行測定,記錄色譜圖。以外標法計算加入水合羥基化頭孢克洛雜質的回收率,同時分別采用外標法與加校正因子的主成分自身對照法對加入的水合羥基化頭孢克洛雜質的含量進行計算并比較結果。
4.4.2 試驗結果
以外標法計算加入水合羥基化頭孢克洛雜質的回收率,結果見表6,水合羥基化頭孢克洛雜質的平均回收率(n=9)為104.03%,RSD為0.82%。

表6 回收率試驗結果Tab.6 The results of sample recovery rate test
分別采用外標法與加校正因子的主成分自身對照法對加入的水合羥基化頭孢克洛雜質的量進行計算,結果見表7。
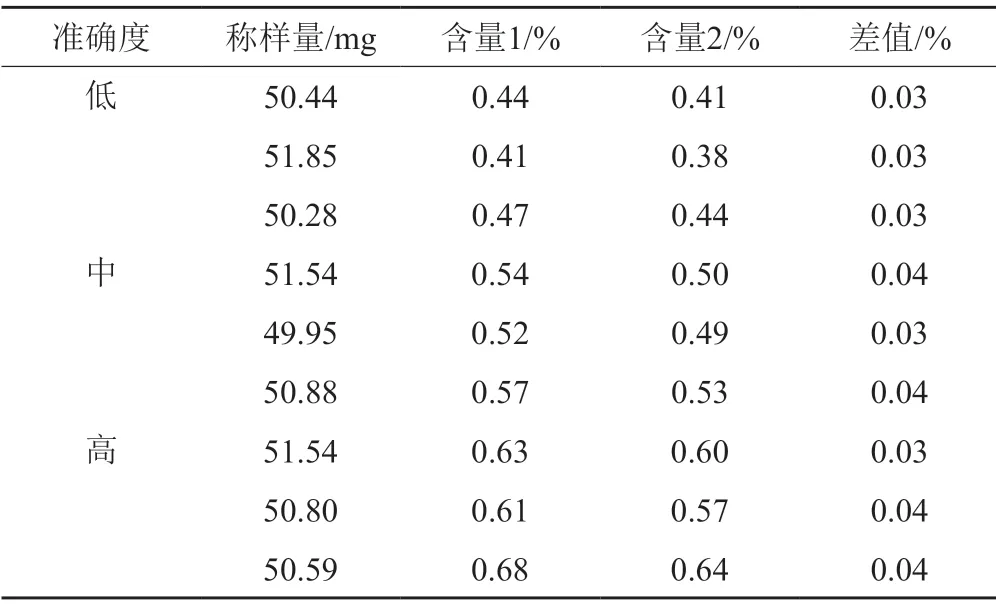
表7 外標法與校正因子法結果比較Tab.7 Comparison of the results of external standard method and correction factor method
4.5 溶液穩定性考察
精密量取“2.3.1”項下對照品溶液5 mL,置25 mL量瓶中,用0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋至刻度,搖勻,分別在4℃條件下放置0、1、2、4、6、8、10、12和24h進樣測定,對照品峰面積的RSD為0.23%。表明水合羥基化頭孢克洛雜質對照品溶液在24h內穩定,結果見表8。

表8 溶液穩定性試驗結果Tab.8 The results of solution stability test
4.6 耐用性考察
由于供試品中水合羥基化頭孢克洛雜質未檢出,為了更準確的測定水合羥基化頭孢克洛雜質,使用加標供試品溶液進行試驗。通過改變檢測波長(215、220和225 nm)、柱溫(20、25和30℃)、流速(1.0、1.2和1.4 mL/min)、流動相B中磷酸鹽與乙腈比例(50:50、55:45和60:40,V/V)、流動相A的pH值(3.8、4.0和4.2),以及使用不同品牌的色譜柱(資生堂 CAPCELL PAK C18AQ色譜柱,4.6 mm×250 mm, 5 μm;Waters Sun fire?C18色譜柱,4.6 mm×250 mm,5 μm;島津ODS-SP色譜柱,4.6 mm×250 mm,5 μm)來考察分析方法的耐用性。
4.6.1 試驗過程
按“4.4.1”項下配制外標法對照品溶液。
精密量取“2.3.1”項下對照品溶液4.5 mL,置100 mL量瓶中,用0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋至刻度,搖勻,作為稀釋劑。
精密稱取頭孢克洛原料藥約50 mg,置10 mL量瓶中,加入稀釋劑溶解并稀釋至刻度,搖勻,作為加標供試品溶液。
精密量取加標供試品溶液1 mL,置100 mL量瓶中,用0.27%磷酸二氫鈉溶液(pH2.5)溶解并稀釋至刻度,搖勻,作為1%對照品溶液。
分別精密量取外標法對照品溶液、加標供試品溶液、1%對照品溶液各20 μL注入液相色譜儀,按“2.2”項下色譜條件進行測定,記錄色譜圖。分別采用外標法與加校正因子的主成分自身對照法計算加標供試品中水合羥基化頭孢克洛雜質的含量。
4.6.2 試驗結果
經試驗,以上色譜條件變化均能滿足系統適用性試驗的要求,對供試品中水合羥基化頭孢克洛雜質的含量測定影響較小,采用外標法與加校正因子的主成分自身對照法計算加標供試品中水合羥基化頭孢克洛雜質含量的結果一致,水合羥基化頭孢克洛雜質的校正因子的耐用性考察見表9~11。水合羥基化頭孢克洛雜質相對保留時間1.10~1.12,且保持穩定,水合羥基化頭孢克洛雜質的相對保留時間耐用性考察見表12。
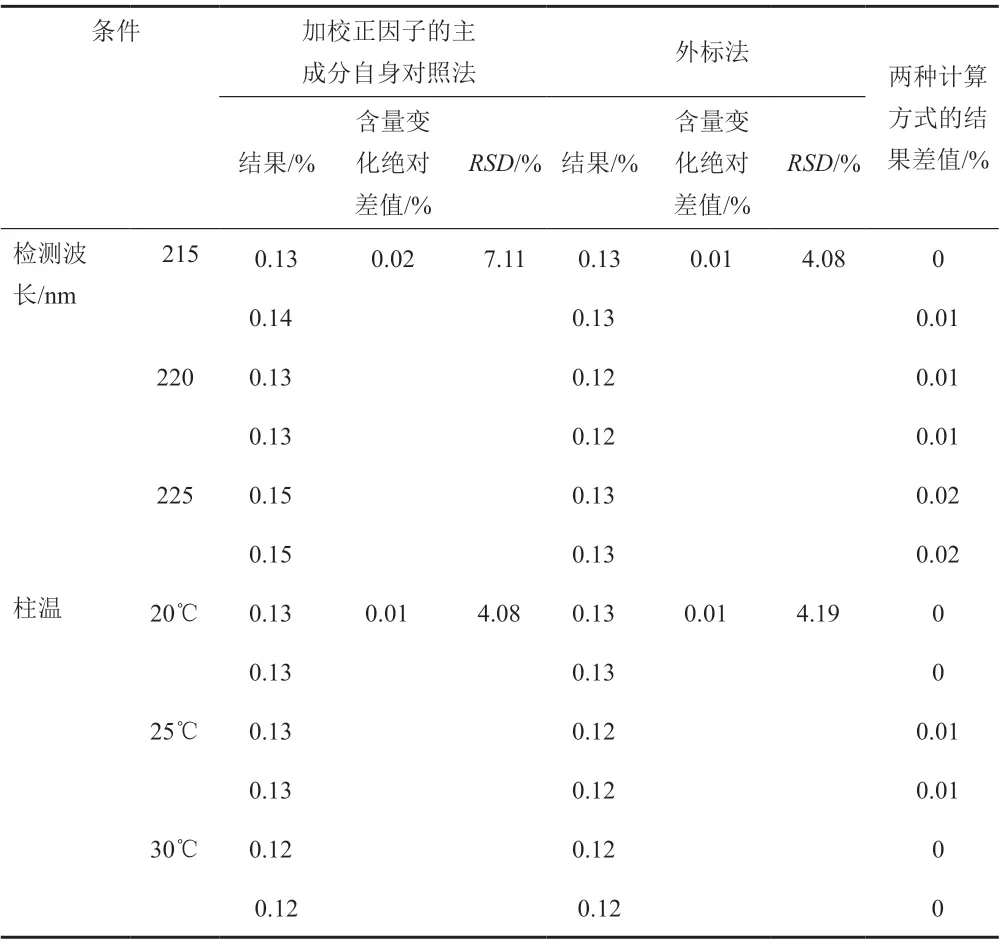
表9 校正因子的耐用性考察-檢測波長和柱溫Tab.9 The correction factor of durability test-detection wavelength and column temperature
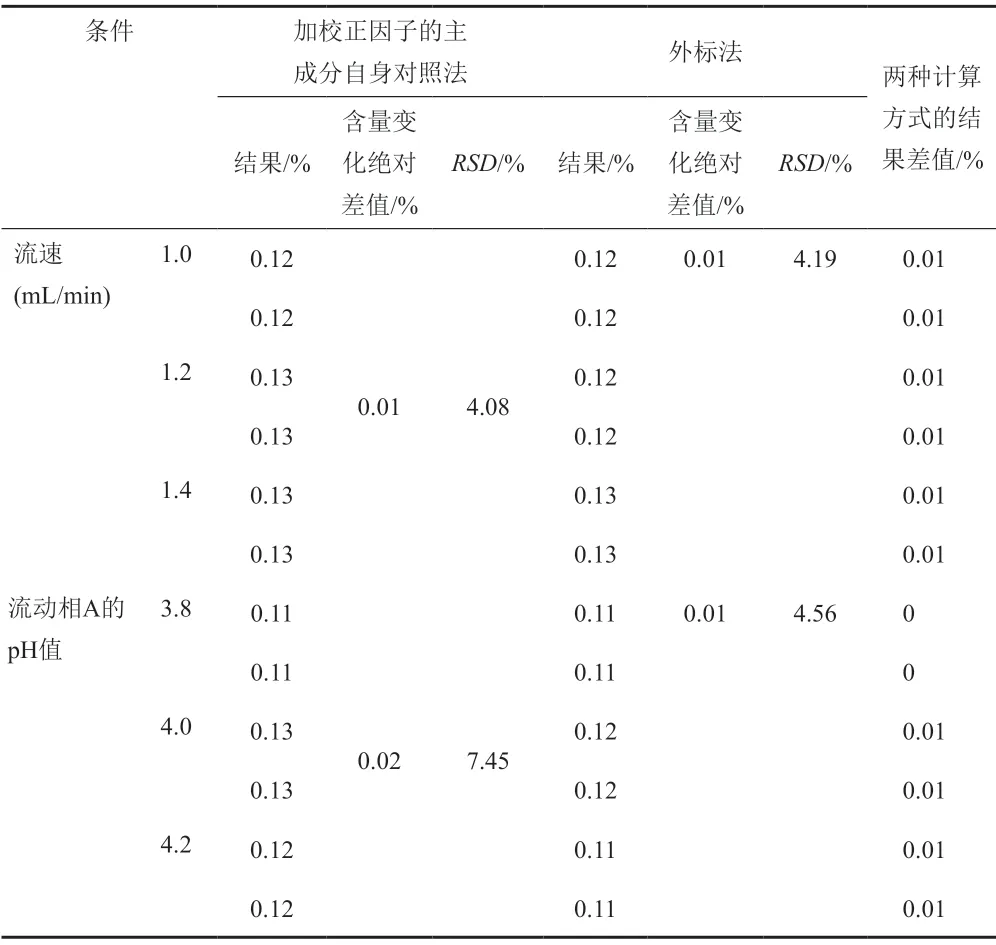
表10 校正因子的耐用性考察-流速和流動相A的pH值Tab.10 The correction factor of durability test- flow and pH of the mobile phase A
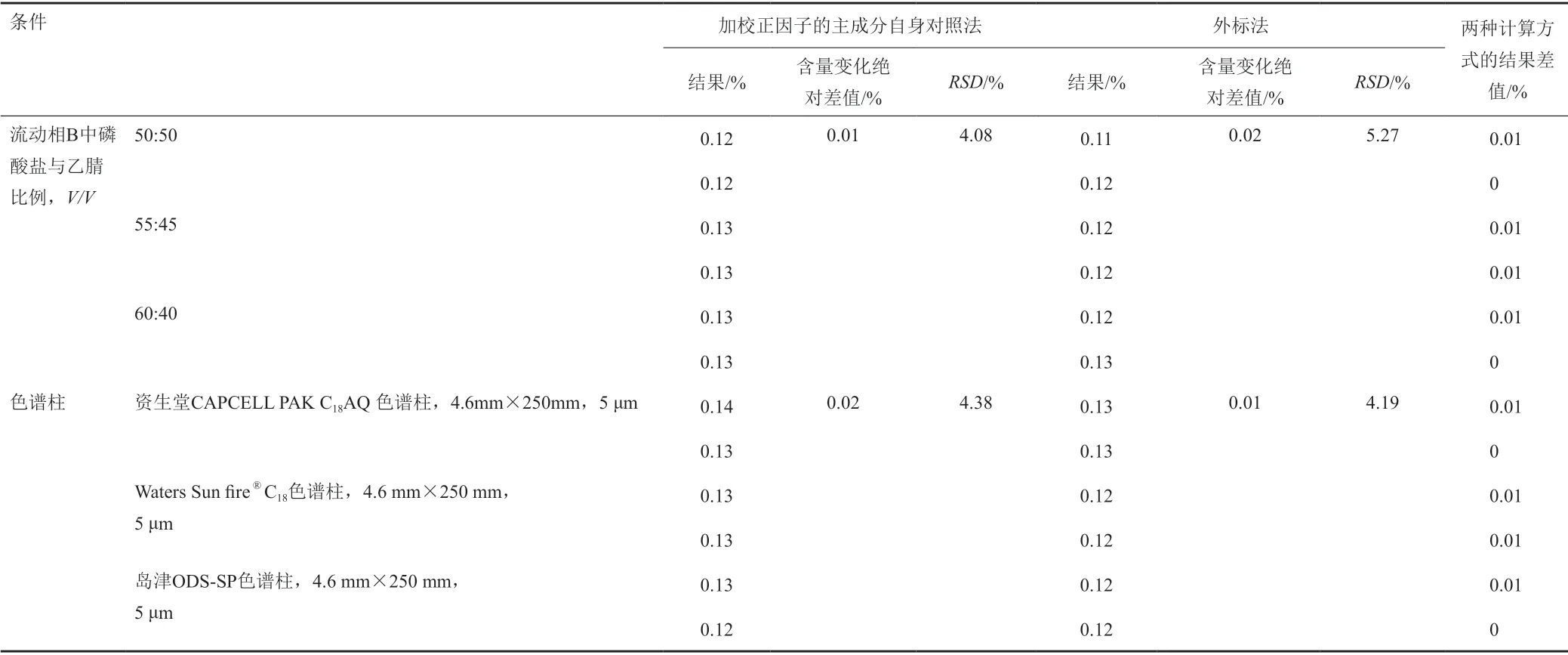
表11 校正因子的耐用性考察-流動相B中磷酸鹽與乙腈比例和色譜柱Tab.11 The correction factor of durability test- the ratio of phosphate to acetonitrile in mobile phase B and column
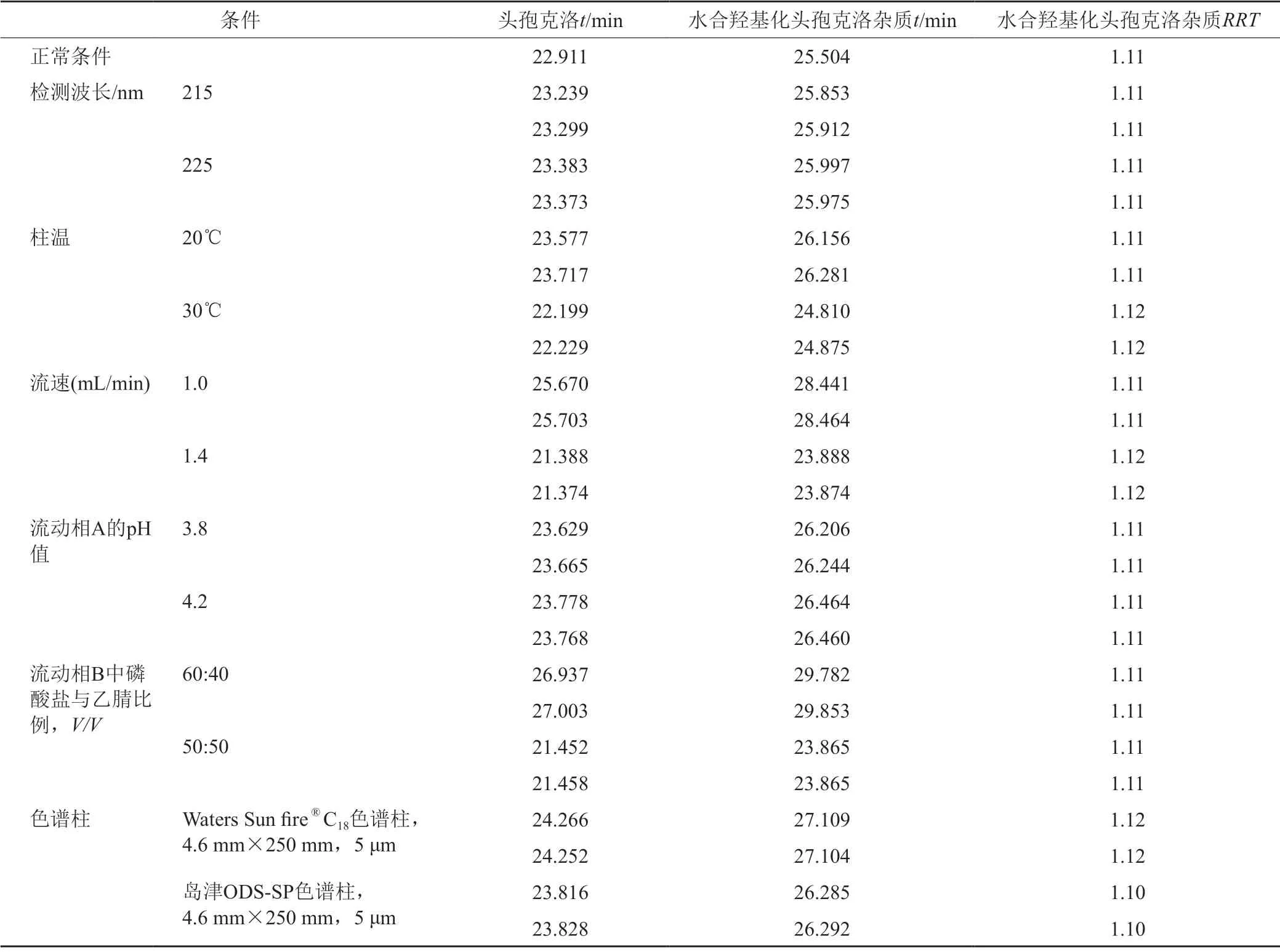
表12 相對保留時間的耐用性考察Tab.12 Relative retention time of durability test
5 討論
5.1 校正因子的確定
本研究通過中間精密度6條曲線,共得出18個校正因子的平均值,確定水合羥基化頭孢克洛雜質的校正因子為1.02。在準確度以及耐用性考察中,將加校正因子的主成分自身對照法和外標法的結果對比,佐證了校正因子的準確性,同時在耐用性試驗中,對該雜質的相對保留時間進行考察,確定該雜質的相對保留時間穩定。
參考ICH關于化藥質量標準中校正因子的指導原則,校正因子在0.9~1.1時,可采用不加校正因子的主成分自身對照法計算含量。本研究中水合羥基化頭孢克洛雜質可不予校正。
5.2 水合羥基化頭孢克洛雜質的控制
水合羥基化頭孢克洛雜質在高溫、強光條件下均會降解產生,在穩定性放置過程中也會降解產生,在頭孢克洛原料藥穩定性放置過程中含量會增加到0.3%左右。頭孢克洛原料藥的質量標準中可以增加對該雜質的單獨控制,采用相對保留時間定位,采用不加校正因子的主成分自身對照法定量研究,以確保原料藥的安全有效。
