重復交叉設計生物等效性研究樣本量計算的確切法與隨機模擬法
2022-01-19 08:40:04王學文袁加盟王洪源
中國衛(wèi)生統(tǒng)計 2021年6期
單 雪 王 登 王學文 袁加盟 王洪源
【提 要】 目的 研究重復交叉設計臨床試驗平均生物等效性評價樣本量計算的確切法與隨機模擬法的統(tǒng)計學特性,推薦選擇較優(yōu)的分析方法。方法 系統(tǒng)的介紹確切法與采用參比制劑校正隨機模擬樣本量計算原理及步驟,采用計算機模擬技術,比較兩種方法在不同情境下的表現(xiàn)。結果 在高變異藥物常見應用情境下,不同參數(shù)設定下通過確切法進行樣本量計算的結果小于采用隨機模擬估計的樣本量,且隨著CVWR%增大,或幾何均值比值偏離1,兩者的樣本量差距增大。結論 確切法與隨機模擬法樣本量計算過程均根據(jù)參比制劑個體內變異(CVWR%)對置信區(qū)間進行放寬,但由于確切法采用的是CVWR%預計值而不是實際取值,且未考慮對點估計值的等效要求等缺點,建議優(yōu)先選擇隨機模擬方法估計高變異藥物等效評價樣本量。
生物等效性(bioequivalence,BE)研究是比較受試制劑(T)與參比制劑(R)的藥代動力學(PK)參數(shù),即評價藥物吸收速度和吸收程度差異是否在可接受范圍內的研究,一般建議采用2制劑2周期交叉設計的方法。根據(jù)受試/參比制劑主要PK參數(shù)幾何均值比值(geometric mean ratio,GMR)的90%置信區(qū)間評價,等效標準為(80.00%,125.00%)[1]。
某些藥物由于吸收、代謝、穩(wěn)定性等原因,導致一個或多個PK參數(shù)的個體內變異系數(shù)(within-subject coefficient of variation,CVW%)大于或等于30%,稱為高變異藥物(highly variable drug,HVD)[2]。對于安全性較好的高變異藥物,PK參數(shù)等效評價采用參比制劑校正的生物等效性(reference-scaled average bioequivalence,RSABE)方法[3]。與2×2設計非校正等效評價的區(qū)別是RSABE等效標準隨CVWR%實際取值變化,需要采用高階的交叉設計,即重復交叉設計,允許在80.00%~125.00%基礎上放寬等效界值,同時要求GMR在80.00%~125.00%內[2]。
目前為止,已有學者發(fā)表了高階重復交叉設計樣本量估算方式,Phani等基于2×2交叉設計的近似樣本量公式[4]、劉甜甜等的確切法[5]、隨機模擬估計方式[6]。本研究將以高變異藥物生物等效性評價試驗為例,詳細介紹重復交叉設計樣本量計算的隨機模擬技術,并與確切法進行統(tǒng)計特性比較。
樣本量估計方法
1.高變異藥物生物等效性評價樣本量計算的確切法
在試驗設計階段,通過既往資料估計參比制劑的個體內標準差SWR,根據(jù)以下公式求得經調整的等效性界值,通過迭代計算相應的2制劑2序列2周期試驗設計的樣本量,再乘以固定系數(shù),得到2制劑3序列3周期部分重復設計或2制劑2序列4周期完全重復設計的樣本量[4-5]。等效界值調整公式如下:
其中,SW0=0.25,為法規(guī)規(guī)定的常數(shù)值。
得到2×2設計下的樣本量后,2×3×3(部分重復設計)樣本量為上述樣本量乘以系數(shù)3/4,2×2×4(完全重復設計)樣本量為上述樣本量乘以系數(shù)1/2。
2.高變異藥物生物等效性評價樣本量計算的隨機模擬方法
決定高變異藥物等效評價樣本量的參數(shù),包括參比制劑的個體內標準差SWR,受試制劑的個體內標準差SWT,受試/參比幾何均值比值GMR,以及1類錯誤率α及2類錯誤率β。采用直接模擬樣本參數(shù)形式產生隨機樣本[7-10],判斷是否成功(即等效),相同預計參數(shù)值的100000個隨機模擬樣本的成功率為統(tǒng)計效能,通過迭代得到預設統(tǒng)計效能的最小樣本量。
具體模擬步驟如下:
(1)設定迭代起始樣本量,按照如下方式生成nsims=100000個隨機模擬樣本,每個隨機模擬樣本主要由以下參數(shù)定義:


表1 與試驗設計相關的E(mse)計算公式


④產生nsims個符合卡方分布的SWR2的隨機數(shù)據(jù):參比制劑個體內均方差SWR2=σWR2×rchi(nisms,dfRR)/dfRR,服從自由度為dfRR的卡方分布,SWR為樣本取值,即每一個隨機模擬情境下或單個樣本的實際取值,σWR為估計的SWR真實值。

(3)符合相應標準則將結論計為“成功”,否則,計為“失敗”,并計算成功的百分比,即為在相應樣本量和參數(shù)設置情況下的統(tǒng)計效能。
(4)若該檢驗效能大于等于設定的目標統(tǒng)計效能,則計算過程結束。否則樣本量加一個最小單位(通常為給藥序列數(shù)的整數(shù)倍,如3序列半重復設計為3,2序列完全重復設計為2),繼續(xù)重復步驟2.1 至2.4直至達到停止標準。
(5)模擬結束,輸出達到目標統(tǒng)計效能的最小樣本量。
模擬評價及樣本量計算結果比較
采用高變異藥物生物等效性評價在實際應用中的常見情境及參數(shù)設置進行隨機模擬實驗。試驗設計采用2制劑3序列3周期,設定檢驗水平為單側0.05,統(tǒng)計效能為80%,不同GMR和CVW估計值對應的隨機模擬估計樣本量和采用確切方法估計樣本量比較趨勢如圖1所示。表2提供了部分具體值,并列出了兩種方法計算的樣本量的差值,同時提供了(0.8,1.25)根據(jù)SWR調整后的等效性界值。可以看到,在高變異(CVW≥0.3)范圍內隨機模擬估計的樣本量均不同程度的大于確切法估計的樣本量,且隨著GMR偏離1,或CVW增大,樣本量差異呈擴大的趨勢。

表2 2制劑3序列3周期部分重復交叉設計隨機模擬樣本量估算與確切方法比較

圖1 在不同GMR的情況下確切法和隨機模擬方法所需樣本量隨CV變化的趨勢圖,CVW=0.3為高變異(CVW≥0.3)標準分隔線
當?shù)刃гu價標準固定時,即非高變異藥物,達到一定統(tǒng)計效能,所需要的樣本量,與GMR和個體內變異的關系呈固定規(guī)律。而采用參比制劑校正方法時,由于評價方法的復雜和等效界值的不確定,在不同GMR時,樣本量并不隨CV單調變化。
法規(guī)要求高變異藥物受試制劑與參比制劑PK參數(shù)幾何均值比值點估計值在80~125內。圖2給出了不同GMR預計值及CV預計值的情況,隨機模擬實際數(shù)據(jù)GMR落在限定范圍外的概率。可以看到,同一GMR預計值的情況下,隨著CV增大,GMR隨機模擬實際值不符合等效要求的概率逐漸增大。同一CV預計值的情況下,GMR預計值越偏離1,GMR隨機模擬實際值不符合等效要求的概率顯著增大。GMR預計值及CV預計值對實際GMR落在限定范圍外的影響具有疊加效應。
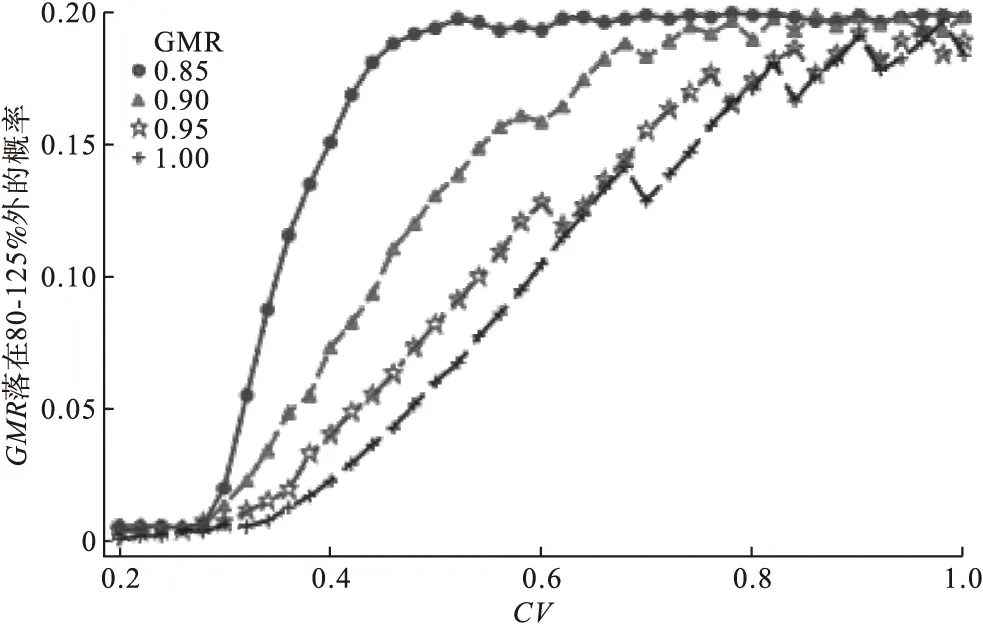
圖2 在不同GMR的情況下隨CV增大GMR落在80~125%外的概率變化趨勢
討 論
高階重復交叉設計由于采用自身對照,可以估計特定制劑的個體內變異,在高變異藥物及窄治療指數(shù)藥物等效評價中得到廣泛應用[11-13]。Phani等[4]提出一種進行間接估計的方法,首先采用SWR計算校正后等效界值,帶入2×2交叉設計的近似公式計算樣本量,然后,2×3×3(部分重復設計)為上述樣本量乘以系數(shù)3/4,2×2×4(完全重復設計)為上述樣本量乘以系數(shù)1/2。2×2交叉設計的樣本量計算公式本身是一種基于較大樣本假設的近似。劉甜甜等[5]采用Meiyu Shen等[14]提出的迭代方式計算確切樣本量的過程替代了公式近似估計2×2樣本量計算過程,然后同樣乘以固定系數(shù)估計重復交叉設計樣本量,對Phani方法進行了一定改進。此外,最近還有學者提出調整參數(shù)后利用PASS軟件的2×2樣本量計算過程進行計算的方式[15],估計結果與劉甜甜提出的方法相似。Laszlo等[6]采用模擬受試者制劑水平數(shù)據(jù)的隨機模擬方式計算樣本量。本文詳細介紹了模擬樣本水平參數(shù)的方式進行樣本量估計的方法,并提供了高變異藥物生物等效性評價實際應用場景下,對確切法及隨機模擬方法的比較。
高變異藥物PK參數(shù)的變異較大,同時生物等效試驗有小樣本的特點,這使得GMR和CVW的不確定性增大,對是否等效起決定作用[16]。確切方法對于生物等效判斷標準進行了過度的簡化。首先,隨著GMR預計值偏離1,CV預計值增大,GMR實際值落在80%~125%以外的概率不能忽視。其次,試驗設計階段預估的SWR往往來自歷史數(shù)據(jù),試驗結束后計算的SWR實際取值存在不確定性,尤其是計劃階段SWR預估值在0.3附近時,有相當?shù)母怕市∮?.294而導致選擇ABE,使得統(tǒng)計效能顯著低于預期,即使選擇RSABE方法,具體等效界值也是不確定的。采用隨機模擬方式計算樣本量,由于隨機模擬SWR的分布,而不是采用固定值,在很大程度上降低了SWR取值不確定的風險。本研究樣本量計算及隨機模擬均采用SAS軟件完成,對不同試驗設計下生物等效評價樣本量計算(FDA及EMA標準略有不同,CDE評價標準同F(xiàn)DA)均已形成網絡操作界面工具。
猜你喜歡
石油瀝青(2021年4期)2021-10-14 08:50:44
藝術啟蒙(2018年7期)2018-08-23 09:14:18
海峽姐妹(2017年7期)2017-07-31 19:08:17
Coco薇(2017年5期)2017-06-05 08:53:16
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
中國教育技術裝備(2015年19期)2015-03-01 02:43:07
俄羅斯問題研究(2012年1期)2012-03-25 09:54:51
體育師友(2012年4期)2012-03-20 15:30:10
