間苯二酚類熱休克蛋白90抑制劑關鍵中間體5-異丙基-2,4-二甲氧基苯甲醛合成研究
2022-01-19 03:03:38薛從建陳秋緣劉巧燕李心忠
福建醫科大學學報 2021年5期
王 薇, 薛從建, 陳秋緣, 劉巧燕, 李心忠, 劉 洋,5
熱休克蛋白90(heat shock proteins,HSP90)是一種ATP能量依賴型蛋白質分子,主要參與調控客戶蛋白的活化、折疊等過程,發揮其分子伴侶的作用。在腫瘤細胞中,HSP90表達高于正常細胞2~10倍,且主要處于活化狀態[1-3]。許多HSP90的客戶蛋白與腫瘤的發生密切相關,如原癌基因絲氨酸/蘇氨酸蛋白激酶、表皮生長因子受體、人表皮生長因子受體-2、血管內皮生長因子、絲氨酸/蘇氨酸蛋白激酶AKT等。通過對HSP90活性的抑制就可同時抑制多種客戶蛋白,從多個信號通路抑制腫瘤,提高靶點藥物的抗腫瘤活性及減少由單一靶點抑制所引起的耐藥性[4-6]。目前研究最多的是間苯二酚類HSP90N端抑制劑。STA-9090和AT13387是該類代表藥物,均已進入臨床[7-8],其合成中均有中間體5-異丙基-2,4-二甲氧基苯甲醛(CAS號1071151-44-4,結構見圖1),而該中間體價格昂貴且需進口。本研究對該中間體的合成工藝進行優化和改進[9-10],為設計合成新型間苯二酚類HSP90抑制劑及降解劑奠定基礎。
1 材料與方法
1.1 材料
1.1.1 儀器 旋轉蒸發儀(RE-5298,上海申生科技有限公司);集熱式恒溫加熱磁力攪拌器(DF-101S,鞏義市予華儀器有限責任公司);超導核磁共振波譜儀(AVANCE Ⅲ,Bruker-BioSpin),液相色譜-質譜聯用儀(6410 Triple Quad LC/MS,安捷倫科技有限公司);數字熔點儀(WRS-1B,上海儀電物理光學儀器有限公司);電子分析天平(BSA-224S,賽多利斯科學儀器有限公司)。
1.1.2 試劑 氫氧化鉀和氧氯化磷(國藥集團化學試劑有限公司);2,4-二羥基苯乙酮和三乙基硅烷(上海達瑞精細化學品有限公司);碘甲烷、三氟乙酸、甲基溴化鎂(上海安耐吉化學有限公司)。以上試劑均為化學純。
1.2 方法 以2,4-二羥基苯乙酮為原料,對2,4位上的酚羥基進行甲基保護,利用格氏試劑甲基溴化鎂將5位酮變成異丙醇,再利用三乙基硅烷和三氟乙酸脫去羥基,最后通過三氯氧磷和N,N-二甲基甲酰胺制成的Vilsmeier試劑引入甲酰基,得到目標中間體5-異丙基-2,4-二甲氧基苯甲醛,合成路線見圖1。
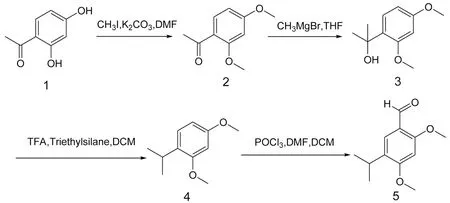
圖1 5-異丙基-2,4-二甲氧基苯甲醛合成路線Fig.1 Synthesis route of 5-isopropyl-2,4-dimethoxybenzaldehyde
1.2.1 化合物2的制備 取100 mL圓底燒瓶,將化合物1[2,4-二羥基苯乙酮(5.00 g,32.89 mmol)]溶于N,N-二甲基甲酰胺(9 mL),加入無水碳酸鉀(9.00 g,65.22 mmol)。將其冷卻至0 ℃,緩慢滴加碘甲烷(6 mL,92.55 mmol)。滴加完畢后升至室溫反應2 h。薄層層析(thin layer chromatography,TLC)監測反應完全后,過濾除去固體,粗濾液用飽和食鹽水洗滌,乙酸乙酯萃取,然后進行濃縮,剩余物用硅膠柱色譜層析分離純化(石油醚∶乙酸乙酯=10∶1),得化合物2共5.85 g,為白色晶狀固體。
1.2.2 化合物3的制備 100 mL雙頸瓶中,加入攪拌子,氮氣保護下將化合物2,4-二甲氧基苯乙酮(5 g,27.75 mmol)溶于四氫呋喃(50 mL)中并冷卻至0 ℃,緩慢滴加甲基溴化鎂的四氫呋喃溶液(12 mL,36.00 mmol,3 mol/L),滴加完畢后緩慢升至室溫反應16 h。TLC監測反應完全后,先后加入水(5 mL)、飽和氯化銨溶液(20 mL),用乙酸乙酯萃取,有機相用鹽水洗滌,用無水硫酸鎂干燥后過濾除去干燥劑,濃縮后硅膠柱層析(石油醚∶乙酸乙酯=10∶1),得化合物3共5.00 g,為無色透明油狀物,產率91.7%。
1.2.3 化合物4的制備 50 mL圓底燒瓶中加入攪拌子,將化合物2-(2,4-二甲氧基苯基)丙-2-醇(3.0 g,15.28 mmol)溶于二氯甲烷(30 mL)中并在冰箱預冷卻至-10 ℃,加入三乙基硅烷(4.50 g,58.60 mmol),緩慢滴加三氟乙酸(6.90 g,63.84 mmol),滴加完畢后緩慢升溫至室溫反應 5 h,TLC點板監測原料轉化完全,停止反應。反應液濃縮后溶于乙酸乙酯,先后用飽和碳酸氫鈉溶液、鹽水洗滌。有機相經干燥,過濾后濃縮干后得粗產物經柱層析(石油醚)得化合物4共2.72 g,為無色透明油狀物。
1.2.4 化合物5的制備 氮氣保護下,將N,N-二甲基甲酰胺(0.43 mL,5.53 mmol)加入二氯甲烷(8.5 mL)并冷卻至0 ℃,緩慢滴加三氯氧磷(0.51 mL,5.59 mmol),滴加完畢后繼續在0 ℃下攪拌反應0.5 h,加入1-異丙基-2,4-二甲氧基苯(0.77 g,4.27 mmol)的二氯甲烷溶液(5 mL),反應溫度升至20 ℃并攪拌3 h,TLC點板監測原料轉化完全后冷卻至0 ℃,緩慢加入水(1 mL)終止反應,濃縮反應液,采用6 N的氫氧化鈉溶液(0.24 g/mL)中和至pH值=7,粗產物用乙酸乙酯萃取,有機相鹽水洗滌,干燥后濃縮,粗產物經過柱層析(石油醚∶乙酸乙酯=10∶1)得化合物5共0.86 g,為黃色固體粉末。
2 結 果
2.1 化合物2的表征 得2,4-二甲氧基苯乙酮(5.85 g),性狀為白色晶狀固體,收率為97.5%。m.p.40.5~41.3 ℃,LC/MS(ESI,m/z):181.1[M+H]+,203.1[M+Na]+。
2.2 化合物3的表征 得2-(2,4-二甲氧基苯基)丙-2-醇(5.00 g),性狀為無色油狀液體,收率為91.7%。LC/MS(ESI,m/z):197.1[M+H]+,219.1[M+Na]+。
2.3 化合物4的表征 得1-異丙基-2,4-二甲氧基苯(2.72 g),性狀為無色透明液體,收率為98.9%。LC/MS(ESI,m/z):181.1[M+H]+,203.1 [M+Na]+。
2.4 化合物5的表征 得5-異丙基-2,4-二甲氧基苯甲醛(0.86 g),性狀為黃色晶體,收率97.7%。m.p. 47.5~47.7 ℃,LC/MS(ESI,m/z):209.0[M+H]+,231.0 [M+Na]+。其核磁譜圖見圖2、3。1H-NMR (400 MHz, DMSO-d6)δ: 10.19 (s, CHO,1H),7.51 (s, Ar,1H), 6.71 (s, Ar,1H),3.94 (s,2OCH3, 6H),3.13 [p,J=6.9 Hz, -CH(CH3)2, 1H],1.13 [d,J=6.9 Hz, -CH(CH3)2,6H]。13C -NMR (100 MHz, CDCl3)δ: 160.47,158.81,156.74,129.75,124.66,115.10, 94.75,55.93,55.45,26.61,22.69。
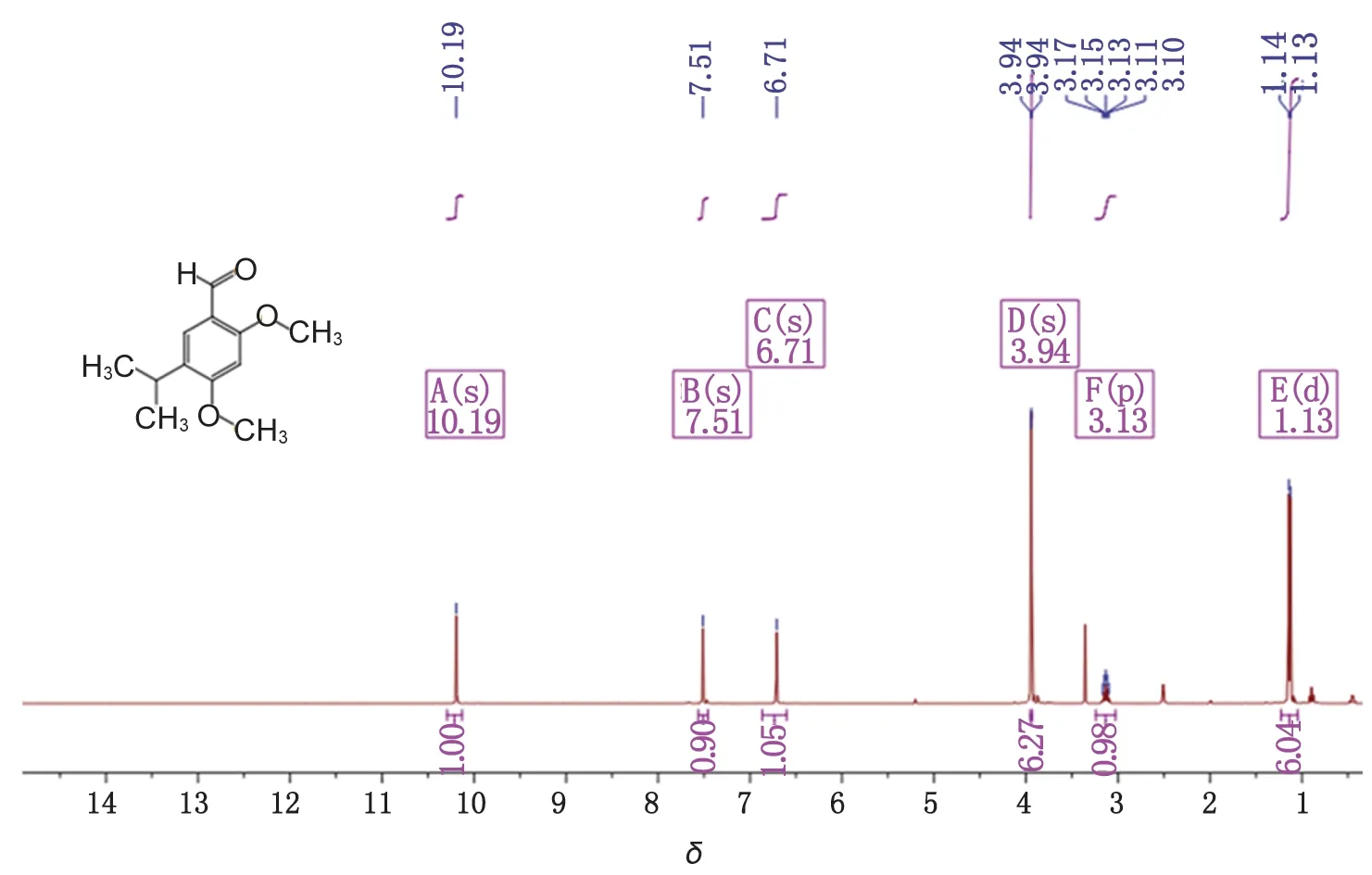
圖2 5-異丙基-2,4-二甲氧基苯甲醛核磁氫譜圖Fig.2 1H NMR spectrum of 5-isopropyl-2,4-dimethoxybenzaldehyde
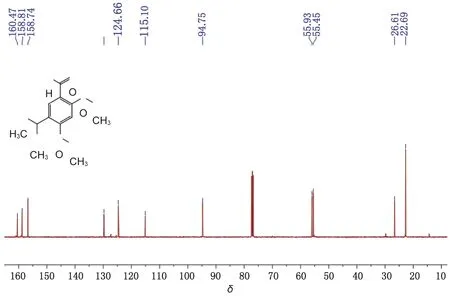
圖3 5-異丙基-2,4-二甲氧基苯甲醛核磁碳譜圖Fig.3 13C NMR spectrum of 5-isopropyl-2,4-dimethoxybenzaldehyde
3 討 論
本研究以2,4-二羥基苯乙酮為原料,用四步合成了HSP90抑制劑中間體5-異丙基-2,4-二甲氧基苯甲醛,總產率達到86.3%。第一步反應2,4-二甲氧基苯乙酮合成因碘甲烷易揮發有毒性,實驗條件改為0 ℃和避光后,收率由原文獻[9]的80%提高到97.5%。第二步格式試劑親核加成反應要嚴格要求做到無水無氧操作,格氏試劑用注射器緩慢加入反應體系中,產率為91.7%,與原文獻99%接近。第三步反應中三乙基硅烷和三氟乙酸按照原文獻的量進行投料后發現產率極低,而將投料量擴大到原文獻的兩倍量后發現原料完全反應,收率由原文獻的93.0%提高到98.9%。第四步反應1-異丙基-2,4-二甲氧基苯與DMF在三氯氧磷作用下進行Vilsmeier-Haack-Arnold甲酰化反應,在5位引入醛基得到目標化合物5-異丙基-2,4-二甲氧基苯甲醛,該反應實際是芳環親電取代反應。由于2位和4位甲氧基的定位效應和空間位阻效應影響,甲酰化產物主要在5位,而不是3位。
結果部分目標產物的核磁氫譜已標注出化學位移對用的氫,10.19單峰為醛氫,7.51 和6.71 兩個單峰為芳環上兩個氫,3.94單峰為甲氧基上6個氫,3.33為水峰,3.13為異丙基上仲氫峰,1.13為異丙基上甲基氫峰。13C -NMR (100 MHz, CDCl3)δ: 160.47為醛基碳,158.81為4位芳碳,156.74為2位芳碳,129.75、124.66及115.10分別為1、6及5位芳碳, 94.75為3位芳碳,55.93和55.45為兩個甲氧基碳,26.61為異丙基上叔碳峰,22.69為異丙基上兩個伯碳,氫譜和碳譜數據與目標產物結構一致。
經過優化的合成路線操作簡單,反應條件溫和,產率高,易純化,適合中試放大生產,為設計合成新型間苯二酚類HSP90N端抑制劑和降解劑奠定了良好的基礎。
