UPLC-MS/MS法同時測定一清顆粒中5種成分的含量
2022-01-17 08:35:14黃麗杰崔燕貞李曉靜
中國民族民間醫藥 2021年23期
岳 磊 黃麗杰 崔燕貞 李曉靜
1.鄭州市食品藥品檢驗所,河南 鄭州 450000;2.鄭州大學第三附屬醫院,河南 鄭州 450000
一清顆粒,作為一種臨床常用的中成藥,在《中國藥典( 2015 版) 》中規定,其處方為黃連、大黃和黃芩這三味藥材,功能與主治為清熱、瀉火、解毒、化瘀、涼血、止血[1]。現行的藥典規定中,在含量測定項下使用高效液相色譜(HPLC)法,僅以黃芩苷作為目標物進行測定,不能有效地針對黃連及大黃進行有效的質量控制。近期關于一清顆粒質量控制的研究集中在制備工藝[2]、指紋圖譜[3]、使用HPLC法檢測更多的目標化合物[4]及針對其中大黃蒽醌類化合物的測定進而對其進行有效的質量控制[5-7]。本文以超高效液相聯合三重四級桿質譜法(UPLC-MS/MS法)來針對一清顆粒中的5種標識成分進行含量測定,質譜檢測法可以在測定多種成分的時,對被測成分在液相條件下的分離度沒有強制要求,能具有更高的專屬性和靈敏度。通過對一清顆粒中黃芩苷、鹽酸小檗堿、大黃素、大黃酚及大黃素甲醚的液相條件和質譜條件進行優化,形成較為成熟的UPLC-MS/MS含量測定方法,可以在日常工作中幫助檢驗檢測人員有效地提高檢驗工作時的工作效率和準確程度,為更全面地針對不同廠家,不同批次的一清顆粒的質量評價和控制提供技術支持。
1 儀器與試藥
1.1 儀器 超高效液相色譜儀,型號:ACQUITY UPLC (美國Waters); 線型離子阱質譜分析儀,型號:4000 QTrap(美國Sciex);電子分析天平,型號:XS205DU(瑞士Mettler Toledo);超聲波清洗機,型號:SB-800DTD(寧波新芝生物科技股份有限公司)。
1.2 試藥 對照品均為中國食品藥品檢定研究院生產,分別為:黃芩苷(110715-201619,93.5%)、鹽酸小檗堿(110713-201814,86.7%)、大黃素(110756-201512,98.7%)、大黃酚(110796- 201922,99.4% )、大黃素甲醚(110758-201817,99.2%);一清顆粒均為河南省及鄭州市2018~2020年度安全監督抽檢樣品;乙腈、甲醇(德國Merck);甲酸(美國Fisher);純凈水(杭州娃哈哈公司)。
2 試驗方法與結果
2.1 色譜與質譜條件 色譜條件 色譜柱:ACQUITY UPLC BEN C18(2.1 mm×50 mm,1.7 μm);流動相:乙腈(A)-水(含0.1%甲酸)(B),梯度洗脫程序(0~3 min,20%→90%A;3~4 min,90%A;4~4.5 min,90%→20%A);流速:0.3 mL/min;柱溫:30 ℃;進樣量:1 μL。
質譜條件 電噴霧離子源(ESI);正、負離子實時切換模式;多重反應監測模式(MRM);源噴射電壓:5500V/-4500V(隨離子模式實時切換);源內溫度(TEM):500 ℃;氣簾氣壓力(CAD):20 psi;渦旋氣壓力(GAS1、GAS2):50 psi;碰撞室氣體:氮氣。黃芩苷等5個成分具體質譜參數見表1。
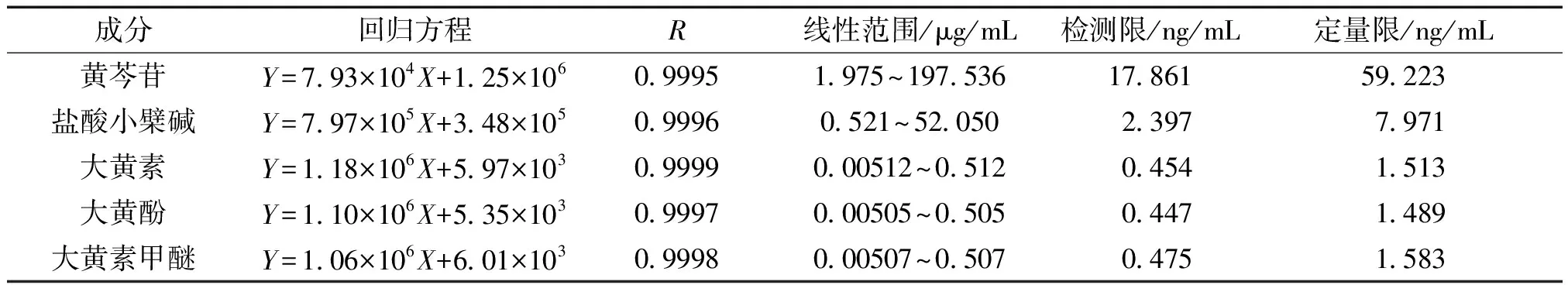
表1 線性關系、檢測限和定量限

表1 黃芩苷、鹽酸小檗堿、鹽酸黃柏堿、腺苷和大黃素的質譜參數
2.2 混合對照品溶液制備 精密稱取5種對照品(黃芩苷、鹽酸小檗堿、大黃素、大黃酚及大黃素甲醚)適量,分別置于適合的容量瓶中,分別取適量體積置100 mL量瓶中,加甲醇制成混合對照品溶液,對應各濃度分別為197.5362μg/mL、52.0497 μg/mL、0.5116 μg/mL、0.5053 μg/mL、0.5071 μg/mL。
2.3 供試品溶液制備 取本品10袋,倒出混合后精密稱定,研細,精密稱取約0.l g,置100 mL錐形瓶中,加入70%甲醇50 mL,精密稱定。置超聲機中滿功率超聲30 min,取出放至室溫,稱重后如有重量損失,使用70%甲醇補足其減失的重量,搖勻后使用0.22 μm微孔濾膜濾過,即得。
2.4 陰性對照溶液制備 取空白輔料,按藥典一清顆粒制法項下的比例混合,制得陰性對照樣品,按2.3項下的方法制得陰性對照溶液。
2.5 專屬性試驗 分別精密吸取稀釋后的混合對照品溶液(精密量取混合對照品溶液5 mL,置 50 mL 量瓶中,加適量甲醇稀釋,并定容至刻度,即得)、供試品溶液及陰性對照溶液各 1 μL,按“2.1”項下的色譜條件與質譜條件進行進樣測定,結果見圖1。結果顯示:供試品及對照品溶液在各成分的提取離子流圖(XIC)中出峰時間一致,而陰性對照溶液在對應的XIC圖中均沒有干擾,其專屬性強。
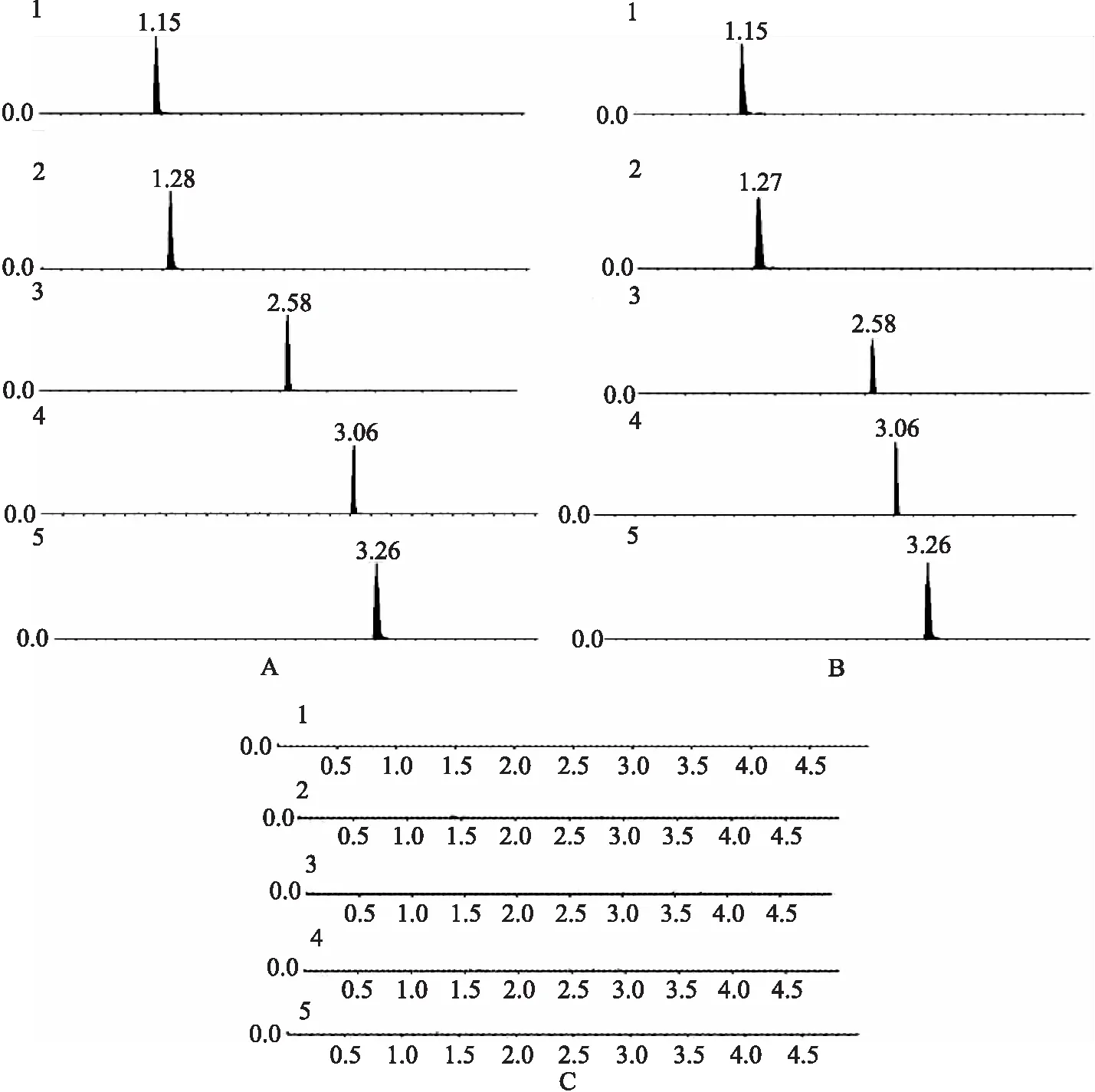
1.黃芩苷;2.鹽酸小檗堿;3.大黃素;4.大黃酚;5.大黃素甲醚;A.對照品溶液; B.供試品溶液; C .陰性對照溶液圖1 5種成分的提取離子流圖
2.6 線性關系考察 精密量取“2.2”項下的混合對照品溶液1.00、5.00、10.00、50.00 mL,分別置100 mL量瓶中,用甲醇稀釋并定容至刻度,搖勻即得。以各化合物的質量濃度為橫坐標(X),以峰面積為縱坐標(Y)進行線性回歸,分別以信噪比S/N=3、S/N=10時的濃度作為檢測限和定量限,回歸方程及相關系數見表1,各成分在對應范圍內的線性關系良好。
2.7 精密度試驗 取“2.5”項下的稀釋后的混合對照品溶液,取連續進樣6次的數據分析。結果顯示,黃芩苷、鹽酸小檗堿、大黃素及大黃素甲醚峰面積的RSD分別為1.37%、2.21%、4.33%、4.49%、4.25%,顯示本方法的精密度符合要求。
2.8 重復性試驗 取編號為01的一清顆粒細粉各6分,平行制備6份供試品溶液,進樣測定。測得各成分的平均含量分別為1.377、0.373、0.762、16.361 、4.357 mg/片,其RSD分別為3.89%、4.18%、4.76%、2.32%、2.18%,顯示本方法重復性符合要求。
2.9 穩定性試驗 取同一份編號為01的供試品溶液,在室溫下放置0、2、4、6、8、10、12 h后測定。結果各成分的RSD分別為2.12%、2.38%、4.17%、4.42%、3.97%,表明該供試品溶液在12 h內具有較好的穩定性。
2.10 加樣回收率試驗 隨機取同一批一清顆粒(編號為01),精密稱取其顆粒細粉0.05 g,精密加入混合對照品溶液適量,制備6份加標供試品溶液的平行樣,計算加樣回收率,結果顯示,各成分的平均加樣回收率分別為96.37%、95.15%、89.57%、90.64%、87.13%,其RSD分別為2.74%、2.19%、2.44%、2.56%、3.03% 。
2.11 供試品含量測定 全部樣品共9批一清顆粒,按照“2.3”項下方法,分別制備供試品溶液進行分析,通過回歸方程計算其5種成分的含量,結果見表2。
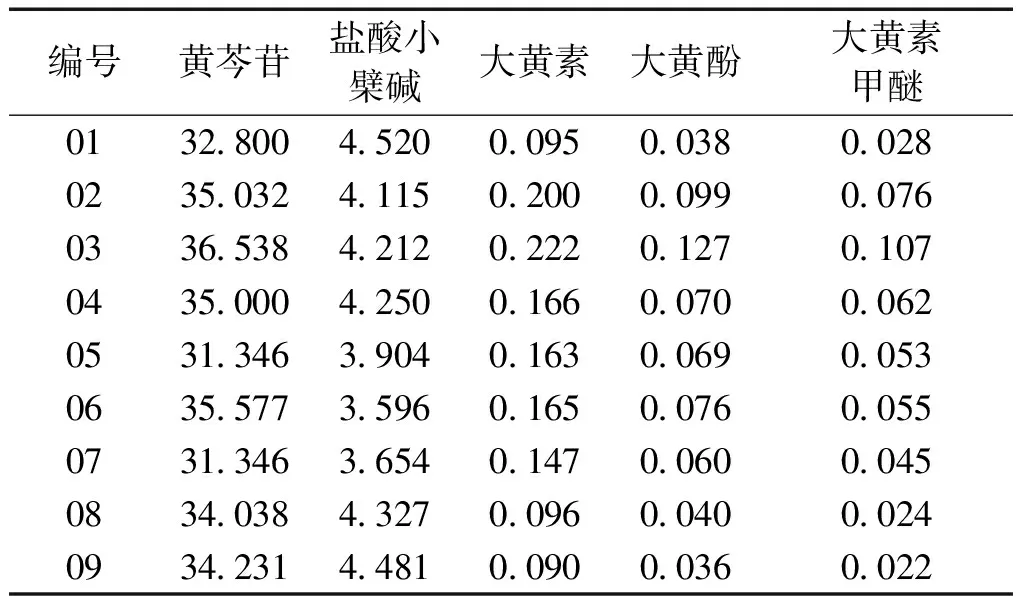
表2 供試品含量測定結果 (mg/包,n=2)
3 討論
3.1 目標化合物的選擇 通過藥典的記載得知,一清顆粒的處方為黃連、黃芩與大黃三味藥材,通過分別煎煮后濃縮成浸膏粉,混合加輔料制成顆粒[1]。其中大黃的用量最大,不使用含量測定的方法對其進行質量控制顯然不夠嚴謹。沿用藥典選擇的黃芩苷作為黃芩的標志物的思路,實驗中增加了鹽酸小檗堿作為黃連的標志物,選用大黃素、大黃酚以及大黃素甲醚作為大黃的標志物進行含量測定,使用液質聯用方法,一次性針對其中的目標成分進行檢測,進而能夠保證對其進行更為全面的質量控制。
3.2 提取方法的優化 針對供試品前處理中的提取方式、選用溶劑與提取時間進行了優化研究。其中提取方式選擇了直接超聲和回流提取,結果顯示二者提取率差異較小,而超聲提取操作較為便捷。選擇的溶劑包括甲醇、50%甲醇、70%甲醇與90%甲醇,最終選用提取率較高的70%甲醇。超聲提取時間在 15 min、30 min與45 min之間進行對比,結果表明選用超聲30 min的樣品,其提取率明顯高于15 min,與45 min的樣品相比差別不明顯。經綜合考量,最終確定選用提取溶劑為70%甲醇,超聲提取時間選擇30 min。
3.3 色譜條件的優化 流動相的優化先采用相同比例的甲醇-水、甲醇-水(含0.1%甲酸)、乙腈-水、乙腈-水(含0.1%甲酸)進行考察。結果顯示使用乙腈作為有機相時,其圖譜的峰型比甲醇的更好,目標化合物的分離程度也更好;而對比加入0.1%的甲酸的水相與純水相,可明顯看到加入0.1%的甲酸可以有效增強這5種化合物的信號強度,也能改善峰形的對稱性。最后考察了不同梯度的洗脫方式,進一步提升了分離效率。最終選擇乙腈-水(含0.1%甲酸)作為流動相,采用梯度洗脫方式進行試驗。
3.4 結果分析 綜上所述,本試驗建立了使用UPLC-MS/MS法同時測定一清顆粒中5個成分的含量,9批樣品測定結果中可以看出,黃芩苷作為藥典方法的控制因素,其含量測定結果顯示,各批次間差異較小,且均符合藥典規定(每袋不得少于21 mg)。鹽酸小檗堿的含量測定結果,各個批次差異也相對較小;而以大黃素、大黃酚、大黃素甲醚作為大黃藥材的特征成分來看,各批次間呈現出較大差異。
