新型可溶性環氧化物水解酶抑制劑TPPU 對缺血性腦卒中的保護機制
2022-01-04 11:27:38黃攀易興陽德陽市人民醫院四川德陽618000
中國老年學雜志 2021年24期
關鍵詞:研究
黃攀 易興陽 (德陽市人民醫院,四川 德陽 618000)
花生四烯酸(AA)是人體內心腦血管活性物質的前體,其代謝產物與腦血管病的發病密切相關〔1〕。AA 的細胞色素P450(CYP)代謝通路是近年研究的熱點〔2〕。AA 在CYP 羥化酶作用下生成羥基二十碳四烯酸(HETEs),在CYP 表氧化酶作用下生成環氧二十碳三烯酸(EETs),EETs 再經過可溶性環氧化物水解酶(sEH)作用下生成生物活性較弱的二羥基二十碳三烯酸(DHETs)。HETEs 具有強力的腦血管收縮作用和促動脈粥樣硬化作用。而EETs 具有擴血管、調節離子通道、抗動脈粥樣硬化等多種生物學功能,對心腦血管具有保護作用。研究顯示AA 的CYP 代謝通路代謝產物(EETs 和HETEs)水平與急性缺血性腦卒中后神經功能惡化密切相關〔3,4〕,提示其在腦缺血性損傷中可能發揮重要作用。sEH 是調控EETs 的關鍵限速酶。研究顯示外周血EETs 降低不僅與缺血性腦卒中后神經功能惡化密切相關,還與腦梗死患者頸動脈狹窄程度和斑塊不穩定密切相關,并受編碼sEH 基因(EPHX2)的調控〔5~7〕。而另有研究表明,EPHX2 基因敲除可增加動脈中動脈阻塞模型大鼠腦血流量,減少梗死體積,對腦缺血具有保護作用,sEH 被認為是缺血性腦卒中防治的新靶點〔8〕。1-三氟甲氧基苯基-3(1-丙酰哌啶-4-基)脲(TPPU)是一種新型的sEH抑制劑,通過抑制sEH 的活性發揮腦血管保護作用,本文對TPPU 發揮的腦卒中保護機制進行綜述。
1 缺血性腦卒中的治療現狀
缺血性腦卒中發病率高,占所有腦卒中的80%左右,溶栓治療和神經保護治療是其兩大主要治療策略。重組組織型纖溶酶原激活(rt-PA)是唯一獲得批準的缺血性卒中的溶栓藥物,其溶栓療效已經在臨床實踐得到證實〔9〕。由于受到時間窗狹窄(發病后4.5 h 內)和顱內出血風險的限制,限制了溶栓治療在臨床上的廣泛應用,因此,神經保護治療一直是缺血性卒中治療領域的夢想和希望。近30 年來,針對腦缺血病理生理環節開發了多種神經保護劑,如抗氧化劑、鈣通道拮抗劑、興奮性氨基酸受體抑制劑和神經營養因子等,全球進行了1 000 多個腦保護實驗研究和100 多個臨床研究,大多在動物實驗有效而臨床試驗無效,導致臨床轉化失敗〔10〕。迄今尚無有效的神經保護劑獲得臨床指南推薦〔11〕,但是人類研發有效神經保護劑的探索一直沒有終止。
2 sEH 抑制劑對腦缺血保護的研究現狀
針對sEH 的神經保護是近年關注的熱點,雖然EPHX2 基因敲除對實驗性腦缺血有保護作用,但EPHX2 基因敲除要用于臨床預防和治療缺血性腦卒中還相當遙遠,因此sEH 抑制劑研發倍受關注。2005 年將sEH 抑制劑12-(3-金剛烷-1-基脲基)-十二烷酸(AUDA)應用在腦缺血神經保護實驗研究中,證實其對實驗性腦缺血有保護作用〔12〕。2007年又證實了另一種sEH 抑制劑轉-4〔4-(3-金剛烷-1-基脲基)-環己氧基〕-苯甲酸(t-AUCB)能改善腦血流,對實驗性腦缺血有保護作用。AUDA 和t-AUCB通過抗凋亡、抗氧化、抗炎、抑制Ca2+內流、線粒體保護、拮抗N-甲基-D-天冬氨酸受體(NMDAR)介導的興奮毒性等多種機制發揮腦保護作用〔13〕。但這兩種傳統sEH 抑制劑從胃腸道吸收差,生物利用度低,要達到有效血藥濃度所用的藥物劑量必須夠大,藥物易在體內蓄積,不良反應較大,動物耐受性差,同時發現這些傳統的sEH 抑制劑半衰期短,體內血藥濃度不穩定,這些原因導致傳統sEH 抑制劑臨床轉化失敗。
3 TPPU 概述
TPPU 是2012 年由加利福尼亞大學獸醫學院分子生物科學中心Bruce 教授等合成的一種新型sEH抑制劑〔14〕。TPPU 分子量為359.3,容易透過血腦屏障與中樞神經系統的sEH 結合,抑制sEH 活性(見圖1)。猴體內藥代動力學表明〔15〕,與傳統sEH抑制劑相比,TPPU 表觀分布容積和濃度-時間曲線下面積大,生物利用度高,半衰期長,1 次/d 即能維持有效血藥濃度,可逆、非競爭性抑制sEH 電流,解離速度快,不會明顯蓄積而影響正常的神經突觸傳導,動物耐受性好。心臟動物模型研究也表明〔16〕,TPPU 可預防心肌梗死后心肌纖維化,具有抗凋亡、抗氧化、保護線粒體等多種功能。
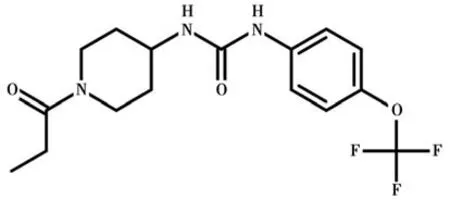
圖1 TPPU 分子結構
4 TPPU 對腦卒中的保護機制
4.1 TPPU 抑制腦卒中后的炎癥反應 炎癥反應不僅可導致腦梗死的發生,而腦梗死的發生也可進一步激活炎癥反應進而形成惡性循環〔17〕。研究顯示腦梗死后炎癥反應便開始進行,可加重腦組織的損傷。白細胞介素(IL)-1 及IL-6 是介導白細胞間相互作用的細胞因子,研究顯示二者可作為評估腦梗死不良預后的獨立標志物,但仍不能超越美國國立衛生院腦卒中量表(NIHSS)評分的價值〔18,19〕,而腫瘤壞死因子(TNF)-α 可由多種細胞產生,其過度表達可引起脂代謝紊亂、胰島素抵抗,加重腦梗死后病情的嚴重程度〔20〕。Tu 等〔21〕通過建立腦梗死小鼠模型后給予TPPU 1 mg/kg 結果發現,TPPU 能明顯促進小鼠神經功能恢復及減少梗死體積及炎癥細胞因子IL-1β mRNA 和TNF-α mRNA 的表達,提示TPPU 可能通過減少梗死后炎癥反應進而促進神經功能恢復。
4.2 TPPU 維持血管穩態作用 血管結構和功能穩態平衡是人體生理活動的基礎,一旦血管穩態發生失衡將導致多種疾病〔22〕。血管損傷是導致血管穩態失衡的原因之一,腦梗死發生后必然造成血管損傷,而血管損傷后血管外膜發生相應的變化。研究顯示在豬冠狀動脈發生球囊拉伸損傷0.5 h后外膜即可檢測到中性粒細胞,而內中膜需要更晚時間才能檢測到;而對大鼠的頸動脈給予球囊拉伸損傷后最早在外膜可檢測到血管內皮生長因子〔23〕。說明血管外膜是血管性疾病的起始部位及積極參與者,是治療血管功能異常的新靶點之一,通過對血管外膜的保護可減少血管穩態的失衡進一步加重進而促進疾病的愈合。AngⅡ相關制劑能導致血管壁增厚、膠原沉積,影響血管外膜的重塑造進而造成血管穩態失衡,而研究者對AngⅡ模型小鼠給予TPPU干預后發現能明顯阻止AngⅡ誘導的血管外膜的損傷,并且進一步體外研究還發現TPPU 可能是通過Ca2+-鈣調蛋白/活化T-細胞核因子(NFATc)3 信號通路影響膠原蛋白的合成發揮作用的,提示TPPU可能是治療血管外膜損傷新途徑之一〔24〕。
4.3 TPPU 保護血腦屏障作用 神經元、星形膠質細胞、血管內皮細胞是構成神經血管單元(NCU)的核心成員;血腦屏障(BBB),包括內皮細胞、基底膜、星形膠質細胞的足突和周細胞是NCU 的核心結構,其中血管內皮細胞間的緊密連接(TJ)是保持血腦屏障完整性的關鍵部位,TJ 主要由跨膜蛋白(claudin、occludin 和JAM)和膜相關蛋白(ZO-1、扣帶蛋白)共同組成。基底膜包繞在腦毛細血管外的膜性結構,周細胞鑲嵌其中,是維持BBB 功能的重要組成部分。基底膜破壞可導致腦血管內皮細胞骨架破壞,影響內皮細胞緊密連接,導致BBB 通透性增加。基底膜由蛋白和多糖組成,能被基質金屬蛋白酶(MMPs)家族降解,腦缺血后MMP9、MMP3 的顯著升高與BBB 破壞密切相關。星形膠質細胞作為中樞神經系統最多的一類細胞,其終足包繞大部分的腦毛細血管,是BBB 的重要組成成分之一。星形膠質細胞膜上有豐富的水通道蛋白(AQP)4 表達,AQP4 是腦脊液重吸收、滲透壓調節、腦水腫形成等生理、病理過程的分子生物學基礎。腦缺血早期主要病理改變是BBB 通透性增加,引起腦水腫,因此,NCU 保護的關鍵是保護BBB,抑制通透性增加,減輕腦水腫〔25〕。研究發現TPPU 能明顯減輕腦梗死后模型大鼠BBB 的損傷,其機制為TPPU 能增加與BBB 有關的連接蛋白ZO-1、occludin 及claudin-5 的表達量。此外,TPPU 還能降低血管內皮細胞的損傷進而減輕BBB 的破壞〔26〕。
4.4 TPPU 增加腦灌注作用 腦梗死發生后灌注量的減少是導致神經功能加重的重要原因,低灌注不僅能刺激局部炎癥反應加劇,還能導致過多的氧自由基被激活進而導致病情加重〔27〕。Hao 等〔28〕通過建立線圈型頸動脈狹窄模型,使用TPPU 干預后發現小鼠的神經功能較對照組明顯改善,進一步基礎研究顯示TPPU 對腦低灌注的神經保護作用可能與激活神經調節素(NRG)1/酪氨酸激酶受體(ErbB)4信號通路有關,并能進一步觸發磷脂酰肌醇-3-激酶(PI3K)-蛋白激酶B(Akt)通路,提示TPPU 可發揮多靶點保護作用,減輕小鼠慢性腦低灌注的神經損傷程度。腦梗死后腦灌注的增加也是導致神經功能惡化的原因之一,因此探索合適的TPPU 治療劑量使二者達到平衡尤為重要。綜上,TPPU 可通過多種途徑干預缺血性腦卒中,可能是治療急性缺血性腦卒中的新途徑、新靶點之一。然而,目前有關于TPPU 治療急性缺血性腦卒中仍處于試驗探索階段,需要更多研究進行論證。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
