復方氨基酸注射液(18AA-Ⅱ)中甲硫氨酸氧化雜質的研究
2021-12-31 03:05:20邵天舒周長明
中國藥科大學學報 2021年6期
邵天舒,周長明,李 輝,郭 雷
(北京市藥品檢驗研究院,國家藥品監督管理局仿制藥研究與評價重點實驗室,北京102206)
復方氨基酸注射液(18AA-Ⅱ)是北京市藥品檢驗所承擔的2019年國家藥品評價性抽驗品種之一。該產品是由18種氨基酸按一定比例配制而成的滅菌水溶液。作為臨床上常用的一類平衡型氨基酸補充劑,主要用于不能口服或經腸道補給營養以及營養不能滿足需要的患者通過靜脈輸注以滿足機體獲取氨基酸合成蛋白質的需要[1-3]。其中,甲硫氨酸作為人體自身不能合成的8種必需氨基酸之一,含量占該產品氨基酸總量的5% 左右。因甲硫氨酸化學結構中的硫元素帶有兩對孤對電子,極易與氧化劑發生氧化反應生成甲硫氨酸亞砜,進一步氧化還可生成甲硫氨酸砜(圖1)。已有的實驗結果表明,上述兩種物質均存在引發肝癌的潛在風險[4-6]:其中甲硫氨酸亞砜可能通過參與DNA甲基化而刺激小鼠肝癌的形成;而甲硫氨酸砜則可通過抑制酶前體降解途徑提高肝癌細胞中谷氨酰胺合成酶的活性,而后者是肝癌的一個重要分子標記物。本品在生產灌裝過程中,若工藝控制不嚴極易混入氧氣,造成產品中存在一定量的氧殘留,可導致甲硫氨酸亞砜及甲硫氨酸砜的產生。
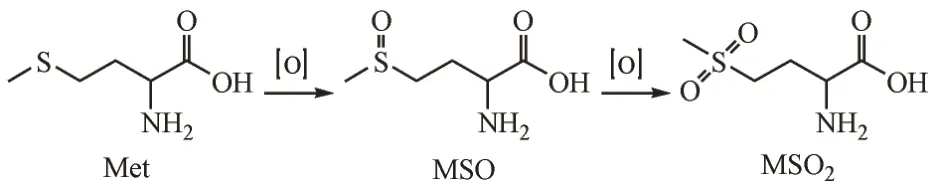
Figure 1 Generation of methionine sulfoxide and methionine sulfone Met: Methionine; MSO: Methionine sulfoxide; MSO2: Methionine sulfone
現行的法定標準《中華人民共和國藥典》二部復方氨基酸注射液(18AA-Ⅱ)品種項下尚未對甲硫氨酸亞砜及甲硫氨酸砜作為特定雜質進行控制[7],造成產品質量存在一定風險。為精準控制產品工藝,提高產品質量,在此次國家評價性抽驗的探索性研究過程中,本研究探索并建立了一種采用鄰苯二甲醛-巰基丙酸溶液為衍生劑,利用液相色譜儀自動進樣器進行柱前衍生,通過C18柱、梯度洗脫對甲硫氨酸亞砜、甲硫氨酸砜與其他氨基酸的衍生物進行分離,并采用高靈敏度的熒光檢測器測定的分析方法,對此次評價性抽驗涉及的國內7 家生產企業共計155 批樣品中甲硫氨酸的雜質含量進行了考察,并對部分企業的產品中甲硫氨酸亞砜含量偏高的原因進行了初步探究。結果顯示,建立的方法在專屬性、線性、準確度、精密度、定量限、檢測限和溶液穩定性等一系列方法學指標均良好,并在一定程度上優于現有的其他測定方法。樣品中甲硫氨酸亞砜含量與樣品殘氧量存在一定相關性,而與抗氧劑的添加量并無明顯關聯。
1 儀器與試劑
Agilent 1260 型高效液相色譜儀(配有自動進樣器及FLD 檢測器,美國安捷倫公司);METTLERXA205 型電子天平(瑞士梅特勒公司);METTLER Seven Easy型酸度計(瑞士梅特勒公司);GS1型頂空氣體測定儀(殘氧量測定用,英國Systech公司)。
甲硫氨酸亞砜(純度:99.9%,批號:BCBZ3966)、甲硫氨酸砜(純度:99.9%,批號:BCBV2699)、鄰苯二甲醛(美國Sigma 公司);復方氨基酸注射液(18AA-Ⅱ)(共涉及國內7家企業(代號分別為B(11批)、F(40 批)、H(8 批)、G(16 批)、L(42 批)、C(1批)、D(37批)),3個規格(總氨基酸含量分別為5%、8.5% 及11.4%),共計155 批,均為2019年國家評價性抽驗樣品);乙腈、甲醇、四氫呋喃均為色譜純;其他試劑均為市售分析純;水為Millipore超純水。
2 方法和結果
2.1 色譜條件
以十八烷基硅烷鍵合硅膠為填充劑(Agilent Poroshell 120 EC-C18,3.0 mm×100 mm,2.7 μm),以醋酸鈉/四氫呋喃溶液(三水合乙酸鈉2.72 g 用適量水溶解,加入三乙胺180 μL、乙二胺四乙酸二鈉0.1 g、四氫呋喃3 mL,混勻加水至1000 mL,用冰醋酸調節pH 至7.2)為流動相A,以醋酸鈉溶液(三水合乙酸鈉2.72 g 加至水200 mL 中,用冰醋酸調節pH 至7.2)-乙腈-甲醇(200∶400∶400)為流動相B;按表1 進行梯度洗脫;流動相流速0.5 mL/min;柱溫40 ℃;熒光檢測器激發波長233 nm,發射波長441 nm;自動進樣器控溫8 ℃;采用儀器自動程序進樣:①吸取硼酸鹽緩沖液5 μL;②吸取衍生劑1 μL;③吸取水0 μL(洗滌進樣針);④吸取樣品1 μL;⑤吸取水1 μL;⑥在空氣中以最大速度混合6次;⑦等待1 min;⑧進樣。
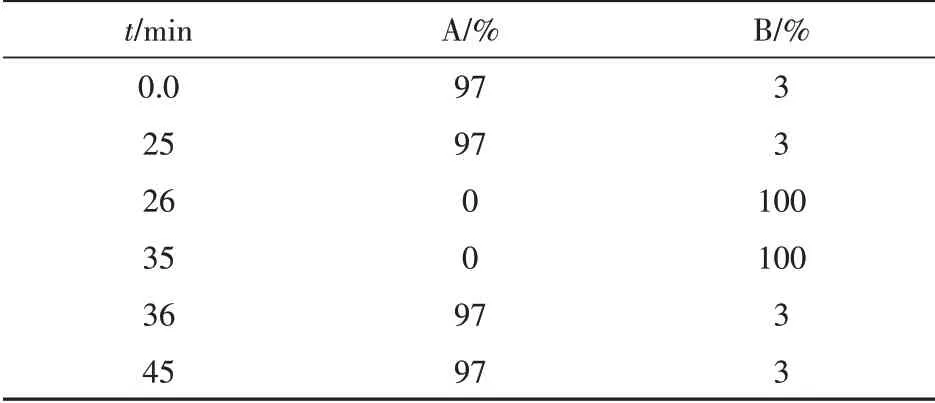
Table 1 Gradient elution program
2.2 溶液的制備
2.2.1 衍生劑(鄰苯二甲醛-巰基丙酸溶液) 取鄰苯二甲醛80 mg,加入0.4 mol/L 硼酸緩沖液(pH 10.2)7 mL、乙腈1 mL、巰基丙酸125 μL,混勻,4 ℃保存。
2.2.2 硼酸鹽緩沖液 取硼酸鈉15.25 g,加水100 mL溶解,用氫氧化鈉溶液調pH至10.2。
2.2.3 對照品溶液 取甲硫氨酸亞砜及甲硫氨酸砜對照品各約50 mg,精密稱定,置100 mL量瓶,加水溶解并稀釋至刻度,搖勻,精密量取1 mL,置100 mL 量瓶,加水稀釋至刻度,制成每1 mL 約含甲硫氨酸亞砜及甲硫氨酸砜各5 μg 的溶液,作為對照品溶液。
2.2.4 供試品溶液 精密量取復方氨基酸注射液(18AA-Ⅱ)樣品2 mL,置20 mL 量瓶中,加水稀釋至刻度,搖勻,作為供試品溶液。
2.2.5 空白對照溶液 除不加甲硫氨酸外,照《中華人民共和國藥典》復方氨基酸注射液(18AA-Ⅱ)處方配制含其余17 種氨基酸及輔料焦亞硫酸鈉的溶液,作為空白對照溶液。
2.2.6 供試品添加對照品溶液 量取復方氨基酸注射液(18AA-Ⅱ)樣品2 mL,置20 mL 量瓶中,并加入甲硫氨酸亞砜和甲硫氨酸砜對照品,加水稀釋至刻度,搖勻。
2.3 結 果
2.3.1 專屬性試驗 取空白溶液、供試品溶液、對照品溶液以及供試品添加對照品溶液,按“2.1”項下色譜條件進樣,結果顯示對照品圖譜中甲硫氨酸亞砜、甲硫氨酸砜峰型良好,兩者分離度為9.52,供試品圖譜中兩者與前后雜峰分離度分別為4.93、2.19,空白輔料無干擾。色譜圖見圖2。
2.3.2 線性關系考察 精密稱取甲硫氨酸亞砜對照品10.25 mg 至100 mL 量瓶中,加水溶解稀釋至刻度,搖勻,作為甲硫氨酸亞砜對照品貯備液(甲硫氨酸亞砜質量濃度:0.1025 mg/mL);精密稱取甲硫氨酸砜對照品10.44 mg 至100 mL 量瓶中,加水溶解稀釋至刻度,搖勻,作為甲硫氨酸砜對照品貯備液(甲硫氨酸砜質量濃度:0.1044 mg/mL);精密量取上述對照品貯備液各5 mL,置同一50 mL量瓶中,用水稀釋至刻度,搖勻,作為混合線性對照品貯備液。分別精密量取上述混合線性對照品貯備液0.25,0.5,2.5,5.0,7.5 mL,置20 mL 量瓶,加水稀釋至刻度,并取混合線性對照品貯備液原液,作為線性試驗溶液。照“2.1”項下色譜條件,以各對照品溶液質量濃度(x)為橫坐標,以相應峰面積(Y)為縱坐標,按最小二乘法計算線性回歸方程。甲硫氨酸亞砜在0.1281 ~10.2500 μg/mL范圍內,甲硫氨酸亞砜溶液的質量濃度與峰面積的線性方程為Y =58.746x- 1.0501,r= 0.9999;甲硫氨酸砜在0.2610 ~10.4400 μg/mL范圍內,甲硫氨酸砜溶液的質量濃度與峰面積的線性方程為Y =47.240x- 1.0157,r= 0.9998。表明兩者在一定的范圍內峰面積與質量濃度線性關系均良好。
2.3.3 檢測限與定量限 取線性試驗用甲硫氨酸亞砜對照品貯備液(甲硫氨酸亞砜質量濃度:0.1025 mg/mL)、甲硫氨酸砜對照品貯備液(甲硫氨酸砜質量濃度:0.1044 mg/mL)各1mL,分別置100 mL 量瓶,加水稀釋至刻度,搖勻。再將上述稀釋后的甲硫氨酸亞砜溶液、甲硫氨酸砜溶液逐級稀釋后注入液相色譜儀測定,至信噪比S/N 作為10∶1 為定量限,S/N 為3∶1 作為檢測限。結果:甲硫氨酸亞砜定量限為0.13 μg/mL,檢測限為0.04 μg/mL;甲硫氨酸砜定量限為0.26 μg/mL,檢測限為0.09 μg/mL。

Figure 2 HPLC chromatograms of system suitability testA:Standard solution; B:Sample solution; C:Blank solution; D:Sample solution with standard.1:Methionine sulfoxide; 2:Methionine sulfone
2.3.4 回收率 精密量取復方氨基酸注射液(18AA-Ⅱ)樣品(批號:1118100305)2 mL,置20 mL量瓶,分別加入甲硫氨酸亞砜及甲硫氨酸砜混合對照品貯備液(含甲硫氨酸亞砜10.25 μg/mL 及甲硫氨酸砜10.44 μg/mL)0.8、1.0、1.2 mL,加水稀釋至刻度,搖勻,各質量濃度分別平行配制3份,分別精密量取上述各對照品溶液,照“2.1”項下色譜條件進樣,記錄色譜圖,按外標法分別計算回收率,結果見表2。
2.3.5 精密度 取“2.2.3”項下混合對照品溶液,照“2.1”項下色譜條件重復進樣6 次,甲硫氨酸亞砜峰面積RSD 為0.36%,甲硫氨酸砜峰面積RSD為0.64%。
2.3.6 重復性 取樣品(批號:1118100305),按照“2.2.4”項下供試品溶液配制方法配制6 份,作為重復性樣品溶液,照“2.1”項下色譜條件進樣測定,按外標法以峰面積計算,重復性樣品溶液中甲硫氨酸亞砜含量的平均值為0.49 μg/mL,RSD 為0.71%;甲硫氨酸砜含量的平均值為0.23 μg/mL,RSD%為1.29%。
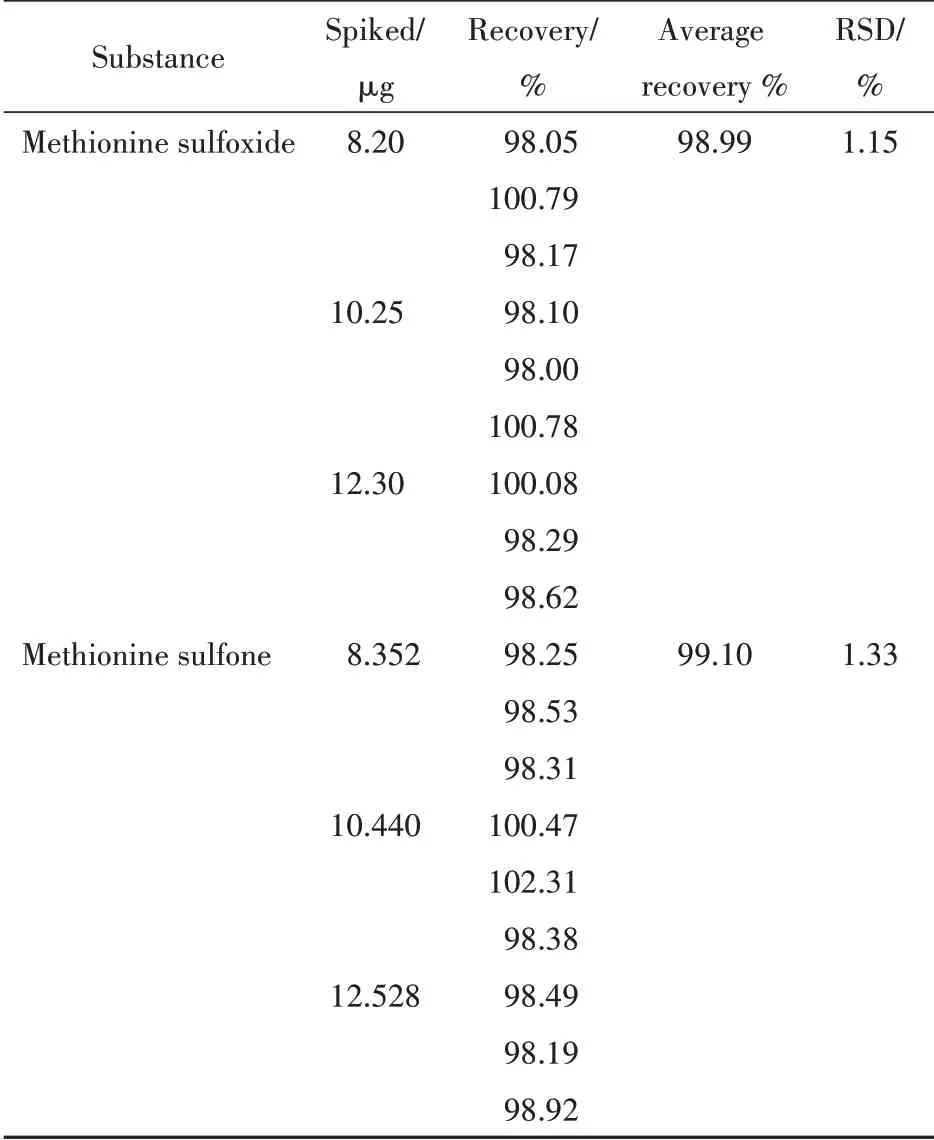
Table 2 Results of methionine sulfoxide and methionine sulfone recovery tests(n = 9)
2.3.7 穩定性 取“2.4”項下供試品(批號:1118100305)溶液分別于0、2、4、8、12、16 h 進樣,照“2.1”項下色譜條件進樣測定,記錄色譜峰峰面積,甲硫氨酸亞砜峰面積RSD 為0.12%,甲硫氨酸砜峰面積RSD 為0.30%,表明供試品溶液在16 h內穩定。
2.3.8 樣品甲硫氨酸亞砜及甲硫氨酸砜的測定結果 取本品,按“2.2.4”項下方法制備供試品溶液,再按“2.1”項下色譜條件進樣測定,對7個廠家的155批樣品進行了檢測,結果見表3。
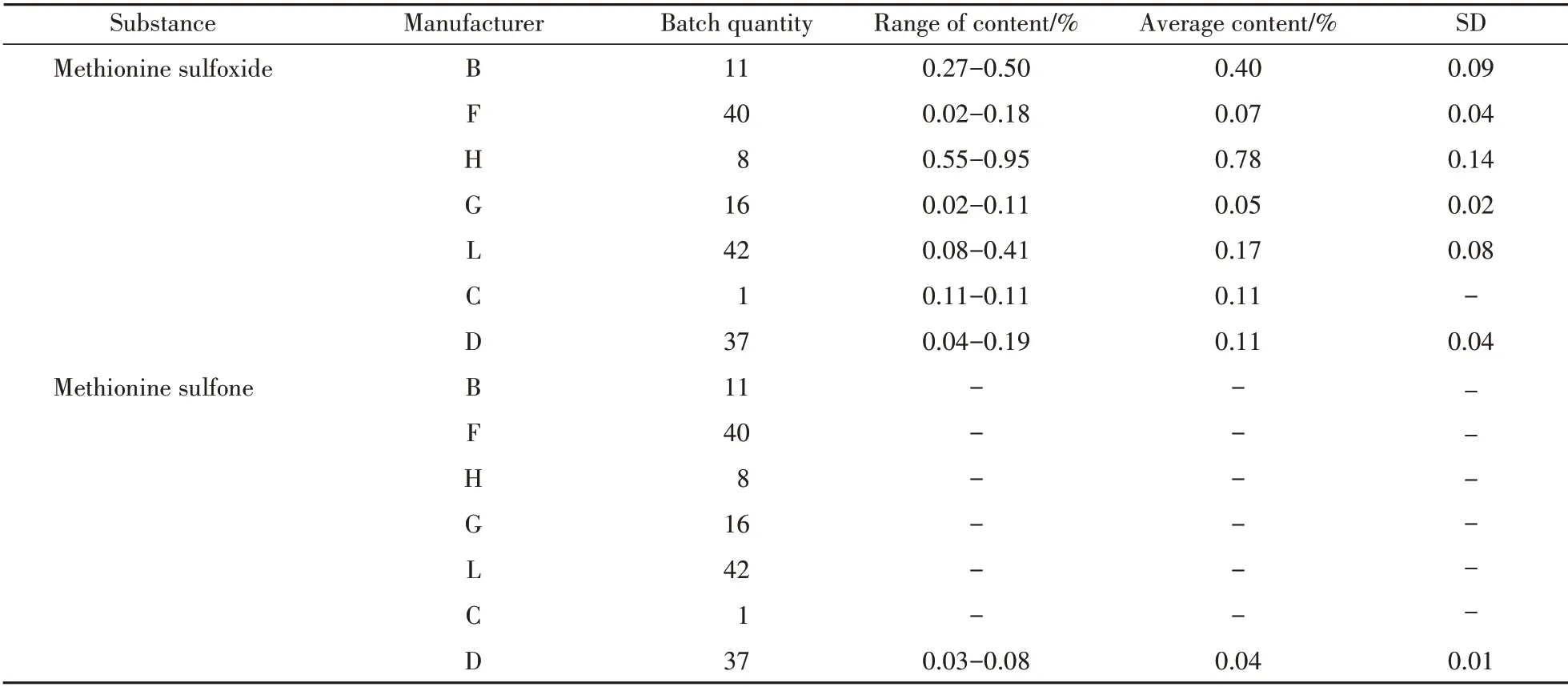
Table 3 Results of methionine sulfoxide and methionine sulfone determination from sample
2.3.9 樣品殘氧量的測定結果 取本品,用頂空氣體測定儀對7 個廠家的155 批樣品上部空間氣體中氧氣比例進行了檢測,B、F、H、G、L、C、D 廠家產品殘氧量均值分別為0.091%、0.053%、0.127%、0.000%、0.065%、0.004%、0.041%。
3 討論
3.1 測定方法的選擇
現有文獻資料中,可同時測定多種氨基酸混合產物中甲硫氨酸亞砜及甲硫氨酸砜的方法有肖玉霞等[8]的氨基酸分析儀法和李莉等[9]的HPLC直接測定法,上述方法雖均有一定的優勢,但也存在諸多不足之處:氨基酸分析儀法需單獨購置特定的儀器,成本較高,且該方法涉及的樣品系明膠水解產物,與復方氨基酸注射液(18AA-Ⅱ)的氨基酸組成存在一定差異,是否適用于本品尚不可知;HPLC 直接測定法為保證甲硫氨酸亞砜及甲硫氨酸砜的分離,需將色譜柱柱溫嚴格控制在4 ℃,因而需配備帶有冷卻功能并精確控溫的柱溫箱,且該方法所用流動相為含有離子對試劑的100% 水相且pH 低至1.9 對C18柱有一定損害,不利于大批量長期測定用,此外因其測定波長選用205 nm的末端吸收波長,在檢測限和定量限方面也不及本方法靈敏。當前,柱前衍生化測定的方法已成為氨基酸類化合物測定的一種常用方法[10-11]。本方法通過采用適當的緩沖體系及流動相梯度洗脫條件,使樣品中的18 種氨基酸衍生產物均在15 min前洗脫,殘余的過量衍生劑在25 min之后洗脫,而待測的甲硫氨酸亞砜和甲硫氨酸砜的衍生產物在此兩者之間的水平基線區段出峰,避免了樣品中的其他18 種組分的干擾,同時選用檢測靈敏度更高的熒光檢測器進行測定。此外,本實驗還針對樣品自動進樣器柱前衍生程序中的關鍵項目鄰苯二甲醛(OPA)用量進行了摸索,分別嘗試吸取OPA 0.5、1.0、2.0 μL。發現當OPA 用量從0.5 μL增至1.0 μL時,相應峰面積亦有所增大;但從1.0 μL 增至2.0 μL 時,不僅相應峰面積不再增大,且在保留時間26 min 左右出現的過量的衍生劑峰明顯展寬,并對樣品中甲硫氨酸砜峰造成了一定干擾,故OPA 用量定為1.0 μL 為宜。全部方法學驗證的一系列指標均符合《中華人民共和國藥典》[12]。
3.2 甲硫氨酸亞砜及甲硫氨酸砜限度的確定
按本研究建立的方法對155 批樣品測定了甲硫氨酸亞砜和甲硫氨酸砜含量,結果發現各批次樣品中均有甲硫氨酸亞砜檢出,不同廠家產品間甲硫氨酸亞砜含量差異較大,H企業產品的甲硫氨酸亞砜含量最高(均值達0.78%);而G企業產品的甲硫氨酸亞砜含量最低(均值僅為0.05%)。另有D 企業的22 批樣品中檢出甲硫氨酸砜。由于《中華人民共和國藥典》該品種項下未涉及甲硫氨酸亞砜與甲硫氨酸砜的控制,而國外藥典亦未收錄此品種,按照復方氨基酸注射液(18AA-Ⅱ)說明書中最大日使用劑量計算,甲硫氨酸每日最大攝入量為7 g左右。參考ICH Q3b新藥制劑中雜質限度的指導原則[13],每日最大攝入劑量在100 mg ~2 g的,雜質限度為0.2%(相對原料藥的百分比,下同);每日最大攝入劑量在2 g 以上的,雜質限度為0.15%,擬定復方氨基酸注射液(18AA-Ⅱ)甲硫氨酸亞砜、甲硫氨酸砜含量均不得過甲硫氨酸標示量的0.2%。依據擬定的標準限度,155 批樣品中有27批甲硫氨酸亞砜含量超出限度,占比17.4%;甲硫氨酸砜含量均未超出限度。
3.3 雜質來源分析
通過對各生產廠家所用甲硫氨酸原料藥的測定,均未檢出甲硫氨酸亞砜及甲硫氨酸砜,表明這兩種雜質均非原料帶入,而是在制劑生產過程中產生的。由于這兩種雜質均是甲硫氨酸的氧化產物,其生成可能與產品生產灌裝過程中混入的氧氣有關。通過7 家生產企業產品殘氧量均值的比較可見,H 企業產品殘氧量整體水平最高,其次是B 企業產品,而H 企業產品甲硫氨酸亞砜含量均值亦最高,B企業產品甲硫氨酸亞砜含量均值排第二位;而產品甲硫氨酸亞砜含量最低的G 企業,其產品殘氧量接近為零。表明甲硫氨酸氧化雜質的產生與產品中殘留氧有明顯關聯。另外,H、B企業生產的產品均為5% 規格,依照藥典處方規定其抗氧劑焦亞硫酸鈉添加量為0.3 g/L,是8.5%、11.4%規格產品焦亞硫酸鈉添加量(0.03 g/L)的10倍,但前者甲硫氨酸氧化雜質的含量均值卻比后者更高,此結果提示添加的抗氧劑是否發揮了預想的作用亦值得商榷。因此,為更好控制產品質量,建議生產廠家進一步優化充氮工藝,同時對不同類型包材對產品殘氧量的影響進行深入研究,以減少甲硫氨酸氧化雜質的產生。同時建議在現行標準的基礎上增訂甲硫氨酸亞砜和甲硫氨酸砜的檢查項目,以對產品質量進行更為有效的控制。
