中藥一致性評價關鍵技術
——定量指紋圖譜聯合 一線多評法研究
2021-12-30 03:13:00閆慧孫國祥孫萬陽蘭麗麗李想蒲道俊胡延雷賈景明陳振鴻沈陽藥科大學中藥學院沈陽00沈陽藥科大學藥學院沈陽00暨南大學藥學院廣州03西南藥業股份有限公司重慶00038國藥集團工業有限公司北京030新疆富沃藥業有限公司新疆阿克蘇地區899
中南藥學 2021年11期
閆慧,孫國祥,孫萬陽,蘭麗麗,李想,蒲道俊,胡延雷,賈景明,陳振鴻(.沈陽藥科大學中藥學院,沈陽 00;.沈陽藥科大學藥學院,沈陽 00;3.暨南大學藥學院,廣州 03;.西南藥業股份有限公司,重慶 00038;.國藥集團工業有限公司,北京 030;.新疆富沃藥業有限公司,新疆 阿克蘇地區 899)
藥物一致性評價,狹義上要求仿制藥與原研藥符合相同的質量標準,廣義上要求兩者具有相同的活性成分和適應證,藥物劑型規格和給藥途徑相同,符合相同的質量標準,達到生物等效,即藥學等效和藥效等效[1]。由于中藥(traditional Chinese medicine,TCM)沒有進行過系統的標準化過程,因此無法采用參比制劑來進行一致性評價。與化藥相比,中藥來源復雜,藥效成分多樣,對某種疾病起效可能為幾種成分的協同作用,故僅檢測其中的一種或幾種成分不能達到整體評價中藥質量的目的,這給中藥的質量評價帶來很大的困難。因此,為了更加系統準確地評價中藥的質量,孫國祥教授建立了中藥定量指紋圖譜評價理論和技術,提出中藥標準制劑控制模式和全質量關控制模式。中藥標準制劑控制模式是中藥一致性評價首選模式,尤其是雙標校正后的標準制劑的對照指紋圖譜將是中藥一致性評價的必由之路。中藥系統指紋定量法從宏觀定性和宏觀定量角度出發,以定量指紋圖譜控制中藥中整體化學物質,進而成為整體評價中藥質量的新方法[2]。本文在中藥標準制劑建立后,采用定量指紋圖譜技術模式即中藥指紋圖譜與多指標成分定量分析相結合的中藥質量控制模式[3]。
但由于中藥成分復雜,某些對照品價格昂貴或難得,使得某些成分的定量控制比較困難,因此,王智民等[4]提出了一測多評法。一測多評法的出現不僅減少了中藥質量評價的成本,也簡化了測定過程,提高了工作效率。一測多評法是通過中藥有效成分間存在的內在函數關系和比例關系,建立樣品中某一有效、價廉、易得的典型成分與樣品其余成分間的相對校正因子(relative correction factor,RCF)以計算樣品中其他成分的量[4]。但是一測多評法在技術上存在4個方面的不足:① 缺乏線性范圍考察和系統方法學驗證技術,導致誤差較大;② 無誤差分析理論和可靠度分析理論,導致分析方法可靠度無法驗證,也無法指導試驗技術人員如何避免誤差;③ 色譜體系無誤差校正方法,通常檢測色譜條件與當初建立標準的色譜系統存在系統定量誤差,誤差傳遞導致最終結果誤差較大(大于5%);④ 理論技術不夠扎實,數學上理論探討不夠深入。
本文在一測多評法的基礎上,提出一線多評法(multi-markers assay by monolinear method,MAML),即基于一測多評法原理并結合中藥定量指紋圖譜理論提出的新方法,彌補了一測多評法在技術上的缺陷和不足。既然中藥定量指紋圖譜承認所確定的全部指紋峰都能整體定量,那么對于主要的幾個指標成分的量值分布必然有確定的函數關系,因此必然能使用一測多評法。一測多評法最大缺陷是忽略了線性范圍,考察濃度范圍太窄,忽視了建立標準系統和檢驗系統之間的量值校正問題即系統定量誤差沒進行任何校正。一線多評法是在多指標定量時采用與指紋圖譜檢測相同的色譜條件同時建立多個活性化合物的標準曲線,利用一測化合物(一測物)和多評化合物(多評物)標準曲線的參數計算校正因子、誤差、可靠度,從而實現用一測物標準曲線對多評物進行定量的分析方法。當方法建立后,以一線多評法對多評物進行定量時的計算公式與一測多評法相同(一般以線性范圍的平均濃度進行試驗),不同的是相對校正因子由多條標準曲線的參數計算而來,具有誤差分析和校正理論及可靠度分析理論。因此一線多評法是在定量指紋圖譜基礎上利用多條標準曲線獲得的與定量指紋系統具有密切關聯度量值的相對校正因子法,由于有雙標校正法而實現定量誤差可控或很小。中藥定量指紋圖譜聯合一線多評能實現對中藥化學指紋物質總量的等位等效控制。 孫國祥教授提出:① 中藥多指標定量分析屬于中藥質量的多點或者多點線質量控制模式(兩點確定一直線,因此是線性控制范圍);② 定量指紋圖譜控制屬于從中藥主組分化學成分出發形成的平面輪廓控制(用指紋圖譜定性控制是形象化外觀圓控制,用中藥指紋圖譜定量控制是對總量的幅度控制-大小不同圓);③ 中藥溶出度控制屬于對中藥主組分指紋溶出的立體控制-球形(實際情況可能為凹凸不規則的球)。中藥質量一致性評價必須是基于標準制劑的點線面立體化控制,見圖1。中藥質量一致性評價的藥學質量研究應從這三個方面開展,才能控制好藥效物質總量的等位,同時控制好體外溶出度一致性來實現對中藥固體制劑工藝過程的一致性控制——質量與藥效實現等位等效。
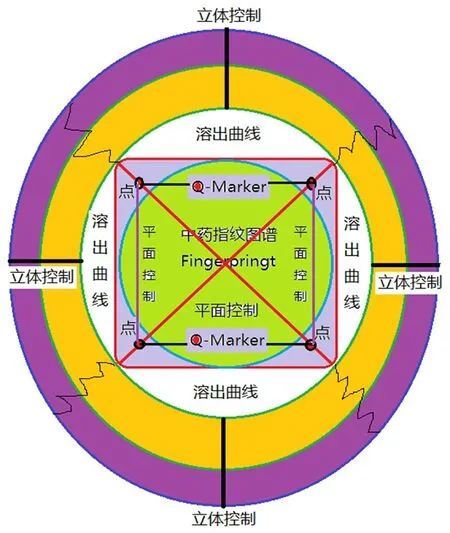
圖1 中藥一致性評價點線面立體控制模型(Q-markers點線控制,指紋圖譜圓面控制和溶出度立體球控制)Fig 1 Dot-line-plane-stereoscopic control model for TCM consistency evaluation(dot-line control of Q-markers,circular plane control of fingerprints and stereoscopic ball control of dissolution)
1 定量指紋圖譜聯合一線多評法模型的建立
1.1 一線多評法線性范圍
在定量指紋圖譜條件下的多指標定量時,必須采用統一化色譜條件同時建立多條標準曲線,用其中一條標準曲線來對多指標分別準確定量的方法稱為一線多評法。一線多評法的最大特點是把標準曲線法最大程度地簡化,節省對照品使用。具體要求為:① RCF(fsi)必須滿足一定線性范圍,推薦10~20倍濃度范圍,最大不超過100倍;② 一測物線性范圍為Rs=[Cs1,Cs2],多評物線性范圍為Ri=[Ci1,Ci2],則被測物濃度應在Ri=[Ci1,Ci2],通常線性范圍的均值濃度為最佳點Cavg=(C1+C2)/2;③ 用標準曲線A=bC+a求RCF更合理(有線性范圍)。用線性方程求RCF特點:① 用最小二乘法計算濃度斜率bi,能準確地揭示峰面積對濃度增長的靈敏度,且給出誤差項ai(標準曲線的截距);② 給出的線性范圍和相關系數能揭示濃度適用范圍和誤差大小;③ 用截距和截距斜率容易計算fsi誤差;④ 方法可靠度能預先準確估算;⑤ 一線多評法方法學驗證后,仍然采用一點法或兩點法對多指標成分定量,因此檢驗工作中沒必要每次都作標準曲線,避免浪費對照品和資源。
1.2 一線多評法應用
一測物S標準曲線和多評物i標準曲線見公式(1)和(2),根據標準曲線計算一測物絕對校正因子fi見公式(3)和(4)。bas=as/Cs,bai=ai/Ci分別為一測物S截距斜率和多評物i截距斜率(即斜率絕對誤差)。可知測定絕對校正因子時,一測物Cs和多評物Ci對bas和bai影響很大,兩者濃度越大則截距斜率越小,即高濃度測定誤差很小。線性平均濃度C0=0.5(C1+C2)(即通常為測定樣品濃度,線性范圍中間濃度)為最佳。用斜率公式直接計算相對校正因子fsi見公式(5),把標準曲線斜率之比稱為簡相對校正因子f'si,當ba=│a/C│≤1%b時可用斜率直接計算fsi,見公式(6)(誤差小于1.0%)。
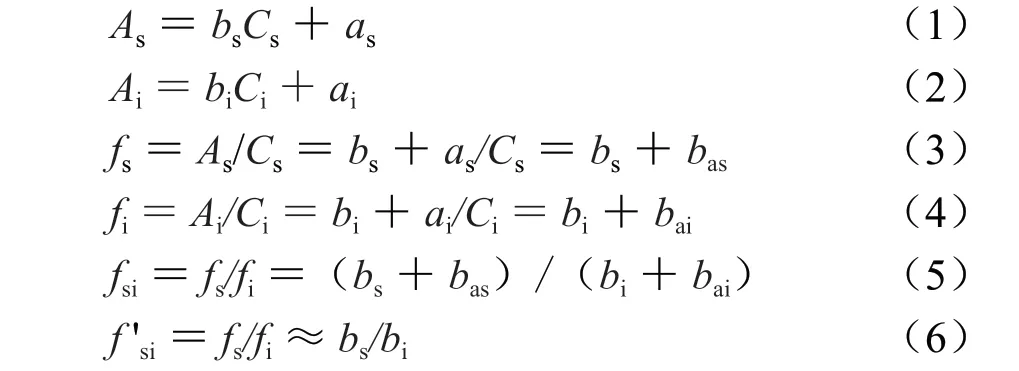
一線多評法是建立在一測物S和多評物i的標準曲線基礎上,具有以下特點:① 線性范圍揭示一線多評法適用范圍;② 給出誤差范圍和可靠度;③ 基于標準曲線的一線多評法科學性和準確性顯著提高;④ 一線多評法只有在精密度和方法重復性均ba=│a/C│≤1%b時,RSD≤2.0%才能保證方法的準確度是可靠的。
1.3 一線多評法誤差
1.3.1 相對校正因子的相對誤差 按誤差傳遞規律得fsi相對誤差見公式(7),主要由一測物S和多評物i截距誤差之差所決定。用均值濃度計算截距斜率時,標準曲線自身誤差的大小決定了相對fsi誤差。計算一線多評的被測物濃度的相對誤差,見公式(8),稱量步驟的相對誤差小是十分重要的,得定量可靠度,見公式(9)。其中被測物相對峰面積誤差代入樣品中i組分精密度的RSDi(REi≈RSDi),一測物S的相對峰面積誤差代入對照品精密度試驗中的RSDs(REs≈RSDs),兩者有部分抵消作用(之差小于1.0%):
① 若△fsi/fsi≤2.0%和△Csi/Cs≤2.0%,則定量誤差小于4.0%;定量可靠度R不低于95.5%;
② 若△fsi/fsi≤1.0%和△Csi/Cs≤1.0%,則定量誤差小于2.0%;定量可靠度R不低于97.7%;
③ 截距斜率誤差大小直接影響定量結果,一測物和多評物濃度越高則截距斜率越小,測定樣品準確度越好。
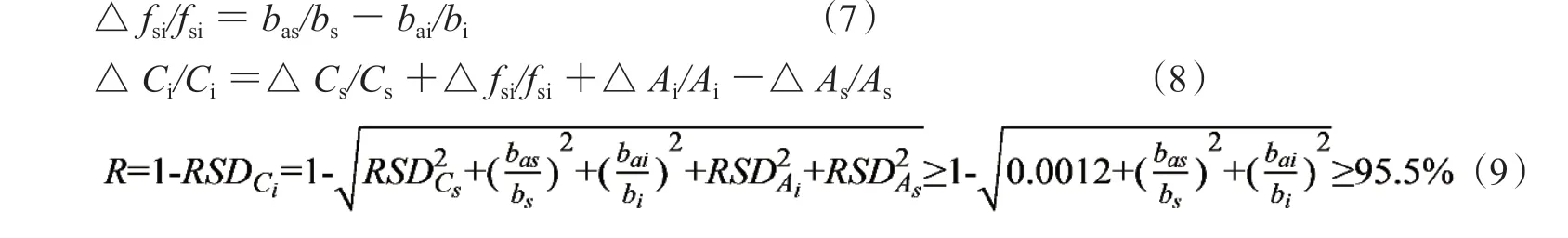
1.3.2 一線多評法的相對誤差 如果標準曲線僅有截距誤差(制備標準溶液的濃度誤差小于1.0%),則測定多評物的可靠度,見公式(9),① 測定S和i峰面積均RSD≤2.0%(不含中間精密度),截距誤差小于1.0%,則方法可靠度大于96.7%;② 若五項相對誤差都小于1.0%,則方法誤差小于2.0%,可靠度大于97.7%,稱量誤差小顯得十分重要;③ 若五項相對誤差都小于2.0%,則方法誤差小于4.0%,可靠度大于95.5%;④ 當斜率誤差較大時方法準確度誤差約5.0%。當只考慮截距誤差時,可靠度估算見表1。因為一線多評法的相對誤差只與測定混合對照品和多組分樣品時的儀器精密度試驗結果和稱量精密度有關。因此一線多評法的方法重復性試驗和準確度試驗是評價該方法整體誤差的必要方法。根據各測量誤差進行不確定度和可靠度大小進行標準劃分,總計有9個等級,見表1。
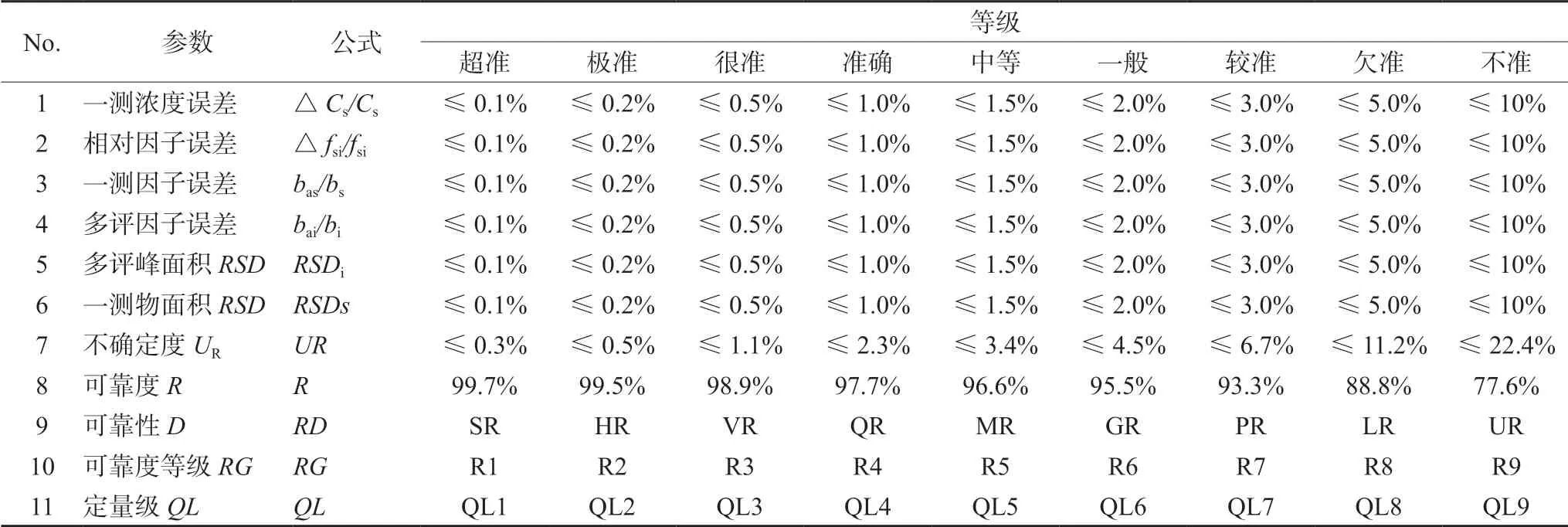
表1 一線多評法的不確定度和可靠度劃分標準 Tab 1 Unreliability and reliability of multi-markers assay by monolinear method
1.3.3 系統指紋定量法原理[5-8]系統指紋定量法(systematically quantified fingerprint method,SQFM)能從宏觀定性角度和宏觀定量角度對中藥進行全方位立體量化控制,適合中藥質量一致性評價。主要包括三個參數:宏定性相似度(Sm),宏定量相似度(Pm)和指紋均化變動系數(α),分別見公式(10~12)。其中α在分級控制時使用較少,只要充分利用好Sm和Pm就能實現對整體主組分指紋的定量控制,該方法還可用于紫外全指紋溶出度評價,同時還能以標準制劑(或參比制劑)為參照對不同批次中藥固體制劑溶出曲線的相似性進行評價,因此SQFM和中藥溶出系統指紋定量法是中藥一致性評價的核心方法,它們的計算方法相同。
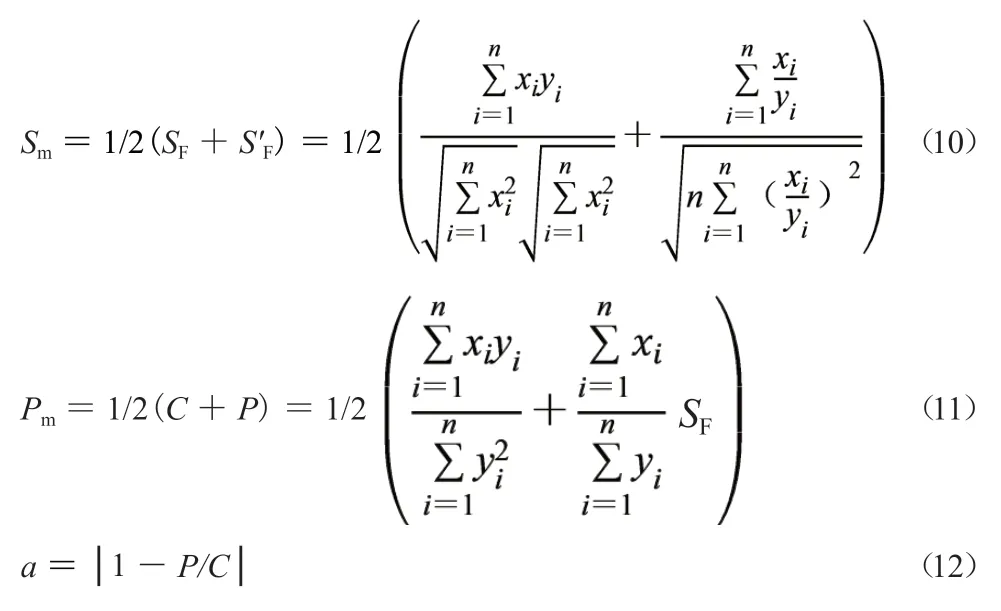
1.3.4 定量指紋圖譜雙標校正法 色譜系統改變會帶來定量誤差,通過雙標校正法進行校正。把建立特征指紋圖譜(標準指紋圖譜)的系統稱為第一色譜系統(first chromatographic system,FCS),對發生誤差后的色譜系統稱為第二色譜系統(second chromatographic system,SCS),在 強 極 性 區 和弱極性區各選擇一個參照物峰作雙標(一般為Q-markers),測定固定濃度雙標混合溶液對應峰面積來計算FCS和SCS的絕對定量校正因子fd1和fdi,見公式(13)和公式(14)[9]。SCS與FCS的絕對定量校正因子之比稱為雙標相對定量校正因子fqi,見公式(15),把FCS定量性質平移到新檢測系統可通過相對定量校正因子fqi與樣品稱樣質量(mi)直接相乘實現校正,見公式(16),稱為雙標校正法[9]。雙標校正法能消除不同色譜系統在不同時間測定的指紋圖譜時所產生的系統誤差,也能保證一線多評法誤差可控或很小。雙標校正法是定量指紋圖譜執行中藥質量一致性評價的必要基礎和有效保證,是一線多評法應用的前提條件。
2 方法
2.1 儀器與試藥

Agilent1100 型液相色譜儀(配有二極管陣列檢測器、四元低壓梯度泵、在線脫氣裝置、自動進樣器),Agilent OpenLAB CDS Chemstation(Edition C.01.07)網絡工作站(Agilent科技有限公司)。Sarturius-BS 110S分析天平(北京賽多利斯天平有限公司);ES-E120D電子分析天平(天津市德安特傳感技術有限公司);超聲波清洗機(深圳市潔盟清洗設備有限公司);安捷倫706ds全自動溶出儀(配有850ds自動取樣器)。
磷酸(色譜純,成都市科龍化工試劑廠);甲醇、乙腈(色譜純,山東禹王和天下新材料有限公司);娃哈哈純凈水(沈陽娃哈哈啟力食品有限公司);庚烷磺酸鈉(色譜純,山東省禹城市中美色譜產品廠);嗎啡(MP,批號:171201-200822)、磷酸可待因(MMP,批號:171203-200504)、甘草苷(LQN,批號:111610-200604)、甘草酸銨(CHAA,批號:110731-201619)和苯甲酸鈉(SB,批號:100433-200301)(對照品,中國食品藥品檢定研究院)。
復方甘草片(S1~S49為廠家A生產;S50~S61為廠家B生產;S62~S73為廠家C生產;S74~S85為廠家D生產;S86~S97為 廠 家E生 產;S98~S109為 廠 家F生 產;S110~S121為廠家G生產;S122~S133為廠家H生產;S134~S145為廠家I生產)。
2.2 溶液的制備
2.2.1 雙標溶液制備 分別取MP和CHAA對照品適量,精密稱定,加甲醇制成每1 mL含200 μg MP和800 μg CHAA的雙標混合對照品溶液,搖勻,即得。
2.2.2 混合對照品溶液制備 分別取MP、LQN、MMP、SB和CHAA對照品適量,精密稱定,加甲醇制成每1 mL含35 μg MP、60 μg LQN、16 μg MMP、160 μg SB和800 μg CHAA的混合對照品溶液,搖勻,即得。
2.2.3 供試品溶液制備 取復方甘草片10片,稱重,研細,精密稱取約4片量,置于50 mL量瓶中,精密加提取溶劑(80%甲醇溶液,含0.5%磷酸)50 mL,精密稱定,45℃超聲處理(功率240 W,頻率40 kHz)10 min,靜置至室溫,再精密稱定,用提取溶劑補足減失的重量,搖勻,0.45 μm濾膜濾過,取續濾液,即得。
2.3 色譜條件
色譜柱為COSMOSIL 5C18-MS-Ⅱ柱(250 mm×4.6 mm,5 μm);以0.2%磷 酸 水 溶 液(含0.005 mol·L-1庚烷磺酸鈉)為水相A,乙腈-甲醇(9∶1,V/V)溶液為有機相B,梯度洗脫(0~10 min,96%~79%A;10~20 min,79%~65%A;20~32 min,65%~47%A;32~45 min,47%~18%A;45~50 min,18%~15%A;50~55 min,15%~96%A);檢測波長為220 nm,柱溫為35℃,流速為1.0 mL·min-1,進樣量為5 μL。
3 結果與討論
3.1 方法學考察
在此定量指紋圖譜系統的色譜條件下,以甘草酸(CHA)峰為參照物峰,并用壓縮因子τ的平方校正理論塔板數后要求應不低于8500[10]。此外,連續進樣測定6次考察儀器精密度;精密吸取S1供試品溶液,分別在溶液制備后的0、2、4、6、14、22 h進樣測定,考察供試品溶液的穩定性;通過分析6個單獨同法制備的S1供試品溶液考察方法的重復性。評價時以CHA峰的保留時間和峰面積為參照,各共有指紋峰的相對保留時間的RSD均小于1.0%,相對峰面積的RSD均小于5.0%,結果表明儀器進樣精密度良好,供試品溶液在室溫放置22 h內穩定,方法重復性良好。
3.2 復方甘草片指紋圖譜建立和評價
按“2.3”項下色譜條件測定9廠家共145批復方甘草片,記錄色譜圖。將積分后*.cdf文件導入【中藥主組分一致性數字化評價系統2.0】軟件(帶審計追蹤功能),以CHA峰的保留時間和峰面積為參照,確定28個共有指紋峰,按平均值法生成標準指紋圖譜(RFP),見圖2,其中10號峰為MP峰,14號峰為LQN峰,15號峰為MMP峰,16號峰為SB峰,27號峰為CHA峰。以此RFP為評價標準對145批復方甘草片進行評價,樣品指紋圖譜與RFP的Sm不得低于0.90,Pm應在80%~120%。根據所得評價結果,以Sm和Pm為參數進行聚類分析,采用SPSS軟件進行系統聚類,結果S75、S81~S85、S107~S109為第Ⅰ類,其余為第Ⅱ類。雖然Ⅰ類Pm數值偏大,但全部在規定范圍內,決定不剔除任何批次樣品。9廠家樣品定量指紋圖譜評價結果見表2(以每個廠家所有批次結果的均值體現),表明9廠家產品質量均合格。其中廠家A生產的批號為20171213、廠家B生產的批號為PDE0712、廠家E生產的批號為3180502、廠家G生產的批號為20180620以及廠家H生產的批號為18053001和18060501的復方甘草片的Sm≥0.95,Pm≈100%,可初步選作復方甘草片的標準制劑[11]。試驗中每隔一段時間進樣測定雙標溶液,記錄色譜圖,計算相對定量校正因子。結果表明在復方甘草片特征指紋圖譜研究期間(約45 d)以及樣品測定期間,fqi始終在0.97~1.03,系統定量性質無顯著變化,不需對色譜系統進行雙標校正。
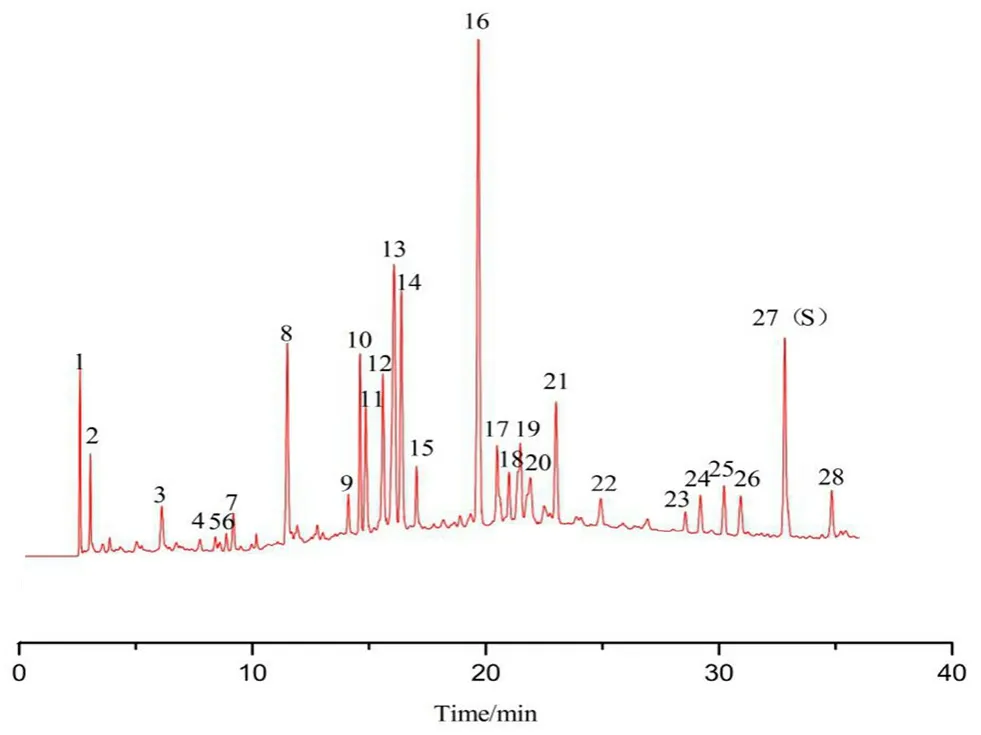
圖2 復方甘草片對照指紋圖譜(RFP)Fig 2 RFP of compound licorice tablet
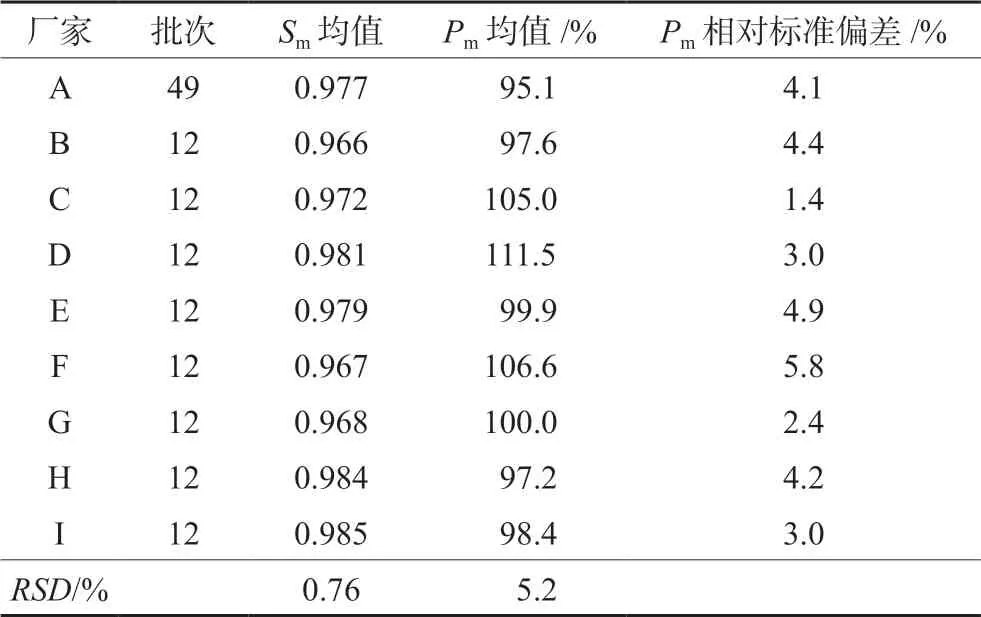
表2 系統指紋定量法評價復方甘草片質量一致性結果 Tab 2 Quality consistency of evaluated by the systematically quantitative fingerprints
從表2的結果可看出,所有廠家樣品的平均Sm均大于0.96,說明不同廠家生產的樣品之間定性相似度均良好,但只以定性相似度進行評價,不同批次間的差異不顯著,不能有效地將不同批次的樣品質量區分開來,只能表明不同廠家樣品之間的化學指紋數量和分布比例十分相似。因此在評價過程中引入Pm是十分必要的,根據Pm結果可進一步區分不同廠家生產的復方甘草片的質量優劣以及同一廠家樣品的批間相似性大小。從結果中可看出廠家A生產的復方甘草片平均Pm值最小,廠家D生產的復方甘草片平均Pm值最大,兩者相差16.4%,說明不同廠家樣品的化學指紋含量存在顯著差異。由此可見,中藥質量評價時僅僅采取指紋圖譜定性參數評價不能達到全面控制中藥質量的目的,定性參數結合指紋圖譜的定量相似度參數才能更準確地評價中藥質量。
3.3 含量測定
本文采用兩種方法測定復方甘草片中5種化合物含量:
① 標準曲線法:精密稱定MP、LQN、MMP、SB和CHAA適量,用甲醇稀釋,制成6個質量濃度的混合對照品溶液。每份溶液在上述色譜條件下各平行測定兩次,以峰面積均值(Aavg)對各對照品濃度(C,mg·mL-1)進行回歸,結果見表3。5種化合物在各線性范圍內的線性關系均很好。利用各標準曲線以外標法計算145批樣品中5種化合物含量,其中CHA含量應在CHAA標準曲線計算所得值基礎上乘以0.9797[12]。
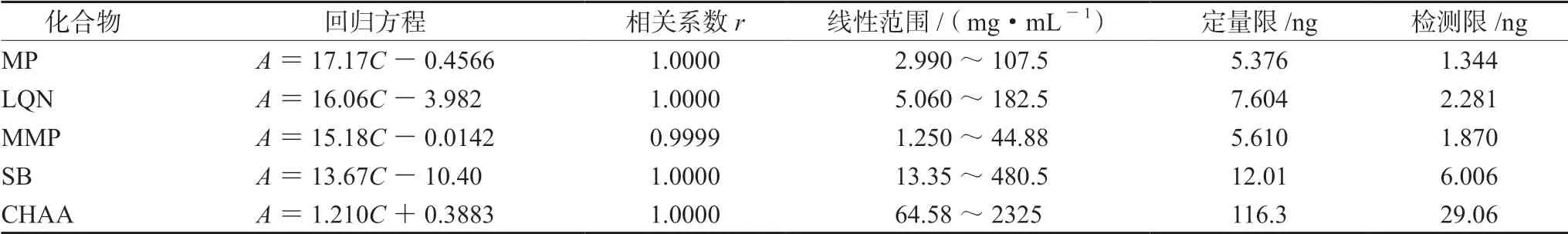
表3 5種化合物線性方程、相關系數、線性范圍、定量限和檢測限 Tab 3 Linear regression equation,correlation coefficient,linearity,LOQ and LOD of the five Q-markers
② 一線多評法:利用標準曲線根據公式分別計算一測物和多評物的bas/bai與fs/fi,進而計算出各標準曲線的fsi,見表4。以標準曲線法計算SB含量,再以SB含量和fsi計算其他4組分含量。另外,各組分的定量可靠度均不低于96.5%,見表4,表明方法可靠性滿足測定要求。以上兩種含量計算方法所得結果見表5(以每個廠家各含量結果均值體現)。另外,《中國藥典》2020年版要求每片復方甘草片中MP含量在0.36~0.44 mg,CHA含量不少于7.3 mg[12],根據表5結果可知,除廠家A和廠家G的MP平均含量不合格外,其余廠家MP平均含量均符合藥典規定,此外,所有廠家CHA平均含量均達到藥典標準。
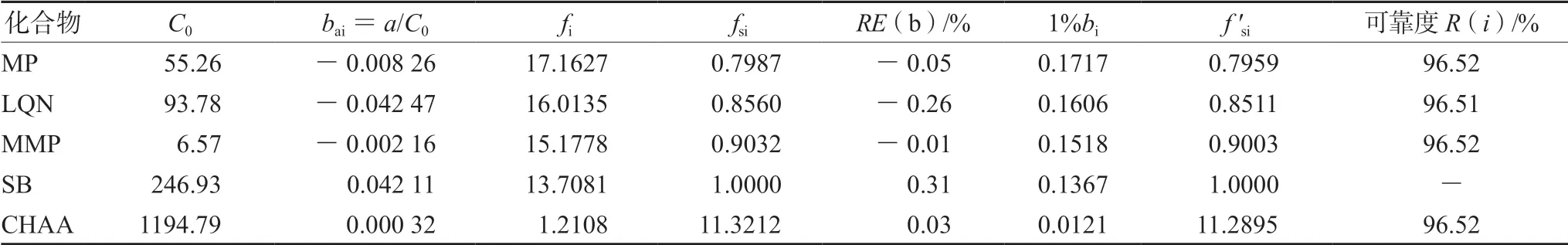
表4 復方甘草片一線多評法的各項參數值 Tab 4 Parameter of compound licorice tablet by multi-markers assay by monolinear method
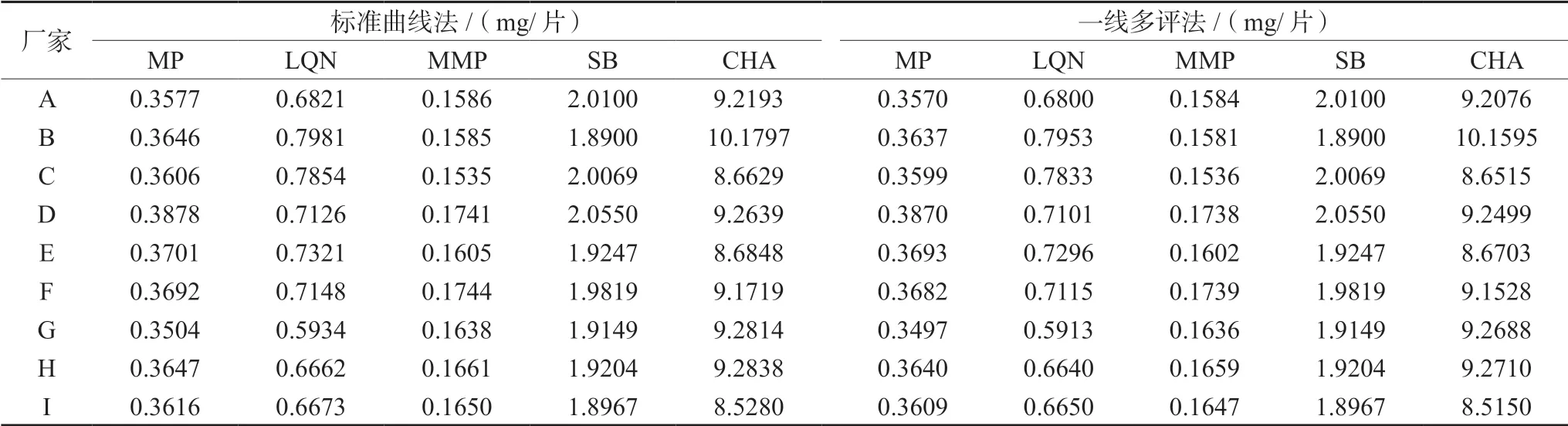
表5 5種化合物含量測定結果 Tab 5 Content of the five Q-markers
將標準曲線法與一線多評法計算所得的每個廠家每種組分的平均含量結果進行比較,計算相對誤差,見表6,兩種計算方法所得相對誤差最大值為0.46%,表明一線多評法能準確測定復方甘草片中5種化合物含量。相較于標準曲線法,一線多評法可節省對照品與分析時間,為中藥及其復方制劑的含量測定提供了一種新的、簡便的思路。
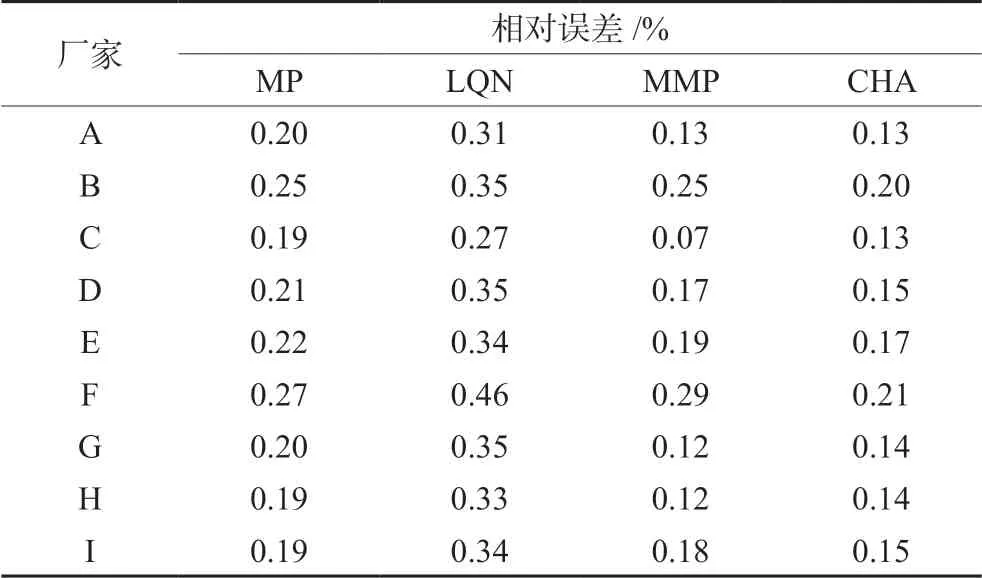
表6 一線多評法與標準曲線法的相對誤差 Tab 6 Relative errors between standard curve method and multimarkers assay by monolinear method
4 討論
本文以復方甘草片為研究對象,通過高效液相色譜法測定145批樣品指紋圖譜,并建立復方甘草片標準制劑的標準指紋圖譜,以系統指紋定量法進行評價。本文提出中藥一致性評價應以定量指紋圖譜聯合一線多評法,實現復方甘草片主組分為中藥指紋組分的復方中藥制劑的整體質量的量化控制。本文提出一線多評法理論、誤差估算方法和可靠度評價方法,該方法主要采用統一化色譜條件,建立雙標校正法,消除測定系統變動帶來的系統定量誤差。通過比較一線多評法和標準曲線法的計算結果,可知一線多評法所得結果準確可靠,操作簡便,節省對照品,可降低檢測成本和減少分析時間,為中藥一致性評價既提供了定量指紋圖譜控制方法,也提供了一線多評法的多指標定量方法,達到了在整體上控制好化學指紋物質一致性的目的。因此,中藥定量指紋圖譜聯合一線多評法構成了中藥一致性評價的首要關鍵技術。本課題組在復方甘草片質量一致性評價中充分使用標準制劑控制模型,用價廉易得的苯甲酸鈉對照品作為一測物來同時定量復方甘草片中其他4種藥效物質,收到理想效果。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中老年保健(2021年4期)2021-12-01 11:19:40
石油瀝青(2021年4期)2021-10-14 08:50:44
中老年保健(2021年4期)2021-08-22 07:08:32
金橋(2020年7期)2020-08-13 03:07:00
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
基層中醫藥(2018年6期)2018-08-29 01:20:20
專用汽車(2016年4期)2016-03-01 04:13:43
中國教育技術裝備(2015年19期)2015-03-01 02:43:07
