HPLC測定羥氯扎胺阿苯達唑復方混懸液的含量及有關物質
2021-12-24 07:46:44張東輝白玉彬申涵露周緒正
中國獸醫學報 2021年11期
張東輝,白玉彬,董 朕,陳 晨,申涵露,李 冰,周緒正
(中國農業科學院 蘭州畜牧與獸藥研究所 農業部獸用藥物創制重點實驗室 甘肅省新獸藥工程重點實驗室,甘肅 蘭州 730050)
羥氯扎胺(oxyclozanide,OXY)又名五氯柳胺,是水楊酰苯胺類抗寄生蟲藥物。作為氧化磷酸化解偶聯劑,其主要作用機理是通過抑制寄生蟲蟲體內的氧化磷酸化過程,來阻止蟲體線粒體中的三磷酸腺苷(ATP)生成,進而導致蟲體因能量代謝紊亂而死亡[1]。OXY具有廣譜、低毒、低殘留等優良特性[2-4],主要用于治療牛羊吸蟲病,尤其對肝片吸蟲治療效果更為突出,同時對絳蟲、線蟲也有良好的治療效果[5-7]。阿苯達唑(albendazole,ABZ)又名丙硫咪唑,是苯并咪唑類驅蟲藥,能選擇性與不可逆性抑制蟲體攝取葡萄糖,使蟲體內源性糖原耗竭,并抑制延胡索酸酶系統,阻礙ATP的產生,致使蟲體死亡,是一種廣譜、高效、低毒的驅蟲藥[8]。
由于牧區養殖環境的復雜性,牛、羊等經濟型動物在養殖過程中往往會出現多種寄生蟲混合感染[9-10],在治療過程中因可選用的復方制劑缺乏,只能選用多種單一驅蟲藥聯合給藥,或者選用廣譜驅蟲藥進行治療。目前國外的復方制劑有羥氯扎胺鹽酸左旋咪唑混懸液,而國內復方制劑只有阿苯達唑伊維菌素預混劑,因此研發一種新的抗寄生蟲復方制劑事不宜遲。本團隊研發的羥氯扎胺阿苯達唑復方制劑可以起到增加抗蟲譜,減少給藥劑量等作用,避免單一驅蟲藥頻繁使用而產生耐藥性問題,有效的促進畜牧業健康發展。李曉婷等[11]采用HPLC只對OXY原料藥中的2個雜質進行檢測,為了更嚴格地控制復方混懸液的質量,本試驗將采用高效液相色譜法建立了羥氯扎胺阿苯達唑復方混懸液的含量及有關物質測定方法,對OXY、ABZ在2015版《英國藥典》中規定的所有雜質進行測定[12-14]。OXY、ABZ及各相關雜質化學結構式見圖1。

圖1 ABZ及相關雜質A~F和OXY及相關雜質G、H的化學結構式
1 材料與方法
1.1 主要儀器Agilent-LC 1290高效液相色譜儀(安捷倫科技有限公司);ME403型電子天平(梅特勒-托利多儀器有限公司);KQ-600DE數控超聲波清洗器(昆山市超聲儀器有限公司);ΜPT-Ⅱ-40L優普超純水制造系統(上海優普實業有限公司)。
1.2 主要試劑OXY標準品(含量質量分數:99.7%,批號:RSB-160703,Dr.Ehrenstorfer GmbH公司);ABZ標準品(含量質量分數:99.9%,批號:100373-201103,中國食品藥品檢定研究院);3,5,6-三氯水楊酸標準品(含量質量分數:99.0%,批號:151708,阿拉丁試劑有限公司);2-氨基-4,6-二氯苯酚鹽酸鹽(含量質量分數:98.0%,批號:A2246,英國藥典委員會);ABZ雜質A、B、C、D、E、F對照品(含量質量分數分別為98.0%,98.0%,98.0%,98.0%,98.0%和97.0%,Trc公司);羥氯扎胺阿苯達唑復方混懸液(含量:4.0%,中國農業科學院蘭州畜牧與獸藥研究所研制)。
甲醇、冰乙酸(分析純,國藥集團化學試劑有限公司);甲醇(色譜純,Merck);甲酸、甲酸銨(色譜純,Flsher);超純水(優普)。
1.3 色譜條件色譜柱:Agilent Poroshell EC-C18(150 mm×4.6 mm,2.7 μm);柱溫:35℃;進樣量:3 μL;流速:0.8 mL/min;流動相:A相為0.1%甲酸-10 mmol/L甲酸銨水溶液,B相為甲醇;檢測波長:292,320 nm;梯度洗脫如表1。

表1 HPLC梯度洗脫條件 %
1.4 溶液制備
1.4.1供試品溶液 取羥氯扎胺阿苯達唑復方混懸液適量(約相當于OXY和ABZ各80 mg),精密稱定,置于100 mL量瓶中,加入適量冰乙酸、甲醇分別超聲10 min,最后再用甲醇定容至刻度,搖勻,即為供試品溶液。
1.4.2對照品溶液 精密移取1.4.1供試品溶液1 mL,置于100 mL量瓶中,用流動相定容至刻度,充分搖勻,即為對照品溶液。
1.4.3空白輔料 按處方比例稱取羥氯扎胺阿苯達唑復方混懸液輔料適量(約相當于OXY和ABZ各80 mg),置于100 mL量瓶中,用流動相稀釋并定容至刻度,搖勻。
1.4.4對照品儲備液 分別精密稱取OXY、ABZ標準品約20 mg及雜質A、B、C、D、E、F對照品各約4 mg,3,5,6-三氯水楊酸約60 mg和2-氨基-4,6-二氯苯酚鹽酸鹽約6 mg置于20 mL量瓶中,用適量DMSO超聲溶解,再用甲醇定容至刻度,作為對照品儲備液Ⅰ。精密移取對照品儲備液Ⅰ1.0 mL,置于20 mL量瓶中,用甲醇定容至刻度,搖勻,作為對照品儲備液Ⅱ。
1.4.5系統適用性溶液 精密稱取OXY和ABZ標準品約1 mg,置于50 mL量瓶中,用冰乙酸酸化的甲醇溶解。精密量取對照品儲備液Ⅱ適量,用甲醇定容至刻度,搖勻,最終配制成主成分與雜質比例為200∶10的專屬性溶液,即為系統適用性溶液。
1.5 專屬性試驗分別取1.4.1供試品溶液、1.4.3空白輔料溶液和1.4.5系統適用性溶液,按1.3色譜條件進樣,記錄色譜圖。
1.6 破壞試驗取羥氯扎胺阿苯達唑復方混懸液適量(約相當于OXY、ABZ各80 mg)5份,置于100 mL量瓶中,加入適量冰乙酸、甲醇各超聲10 min,分別經酸、堿、氧化、光照、高溫破壞后,加甲醇稀釋至刻度,搖勻,作為破壞溶液。破壞試驗操作如下:
強酸破壞:加入1 mol/L鹽酸溶液4.0 mL,室溫條件下放置2 d后,加入1 mol/L氫氧化鈉溶液4.0 mL中和;強堿破壞:加入1 mol/L氫氧化鈉溶液4.0 mL,室溫條件下放置2 d后,加入1 mol/L鹽酸溶液4.0 mL中和;氧化破壞:加入體積分數為3%雙氧水4.0 mL室溫條件下避光放置30 min;光照破壞:置于光照箱內(4 500±500) lx照射2 d;高溫破壞:置于藥物穩定儀中,高溫60℃,放置2 d。
分別吸取上述破壞試驗樣品溶液3 μL,按1.3色譜條件進樣分析,記錄色譜圖。
1.7 精密度進樣精密度:精密量取1.4.5系統適用性溶液3 μL,按1.3色譜條件進樣分析,連續分析6次,計算各個雜質與主成分的保留時間和峰面積的RSD。
中間精密度:精密量取1.4.1供試品溶液6份,由甲、乙2人分別在不同時間段,使用2臺不同儀器,按1.3色譜條件進樣分析,計算各個雜質與主成分的保留時間和含量的RSD。
1.8 定量限與檢測限取1.4.4各雜質對照品及ABZ、OXY主成分對照品儲備液Ⅰ逐級稀釋,按1.3色譜條件進樣分析,記錄色譜圖。以信噪比S/N為3時對應的濃度為檢測限(LOD);以信噪比S/N為10時對應的濃度為定量限(LOQ)。
1.9 線性范圍考察分別精密移取1.4.4下ABZ、OXY以及各雜質對照品儲備液Ⅱ0.10,0.25,0.40,0.50,0.60 mL于5 mL量瓶中,用甲醇定容至刻度,搖勻,即得到不同濃度的線性標準溶液。精密量取3 μL,按1.3色譜條件進樣分析,記錄色譜圖。以峰面積(A)為縱坐標,質量濃度(ρ,mg/L)為橫坐標,按最小二乘法進行線性回歸。
1.10 重復性試驗按1.4.1方法平行制備6份供試品溶液,按1.3色譜條件進樣分析。
1.11 溶液穩定性按1.4.1和1.4.2方法制備供試品溶液和對照品溶液,在室溫條件下放置,分別于0,2,4,6,8,12 h取樣,按1.3色譜條件的進樣分析,計算各雜質保留時間和峰面積的RSD值。
1.12 回收率試驗分別精密稱取OXY、ABZ對照品8 mg,置于10 mL量瓶中,共9份,按處方量稱取空白輔料,加入適量冰乙酸、甲醇分別超聲10 min,精密量取1.4.4雜質對照品儲備液Ⅱ0.8,1.0,1.2 mL,并用甲醇定容至刻度,搖勻,作為低、中、高(80%,100%,120%)3個濃度水平的供試品溶液,每個水平平行制備3份。按1.3色譜條件進樣分析,計算各回收率樣品中雜質的含量(按外標法扣除本底值之后計算)。
1.13 耐用性試驗取系統適用性溶液,在1.3色譜條件下,通過改變流速(±10%)、柱溫(±5℃)、流動性比例(±2%),換用不同類型的不同廠家色譜柱,觀察各組分之間的分離情況以及有關物質含量的變化。
1.14 有關物質含量測定取羥氯扎胺阿苯達唑復方混懸液3批樣品(批號:20201007、20201008、20201009),按1.4方法制備供試品溶液和自身對照品溶液,按1.3色譜條件進樣分析。采用不加校正因子的主成分自身對照法計算雜質含量,記錄供試品溶液和自身對照品溶液色譜圖至主成分保留時間的2倍,其中供試品溶液中任何小于自身對照品溶液主峰面積的0.05倍的峰可忽略不計。
2 結果
2.1 專屬性試驗由圖2可知,各雜質與主成分OXY和ABZ之間以及各雜質之間的峰分離度均大于1.5,且峰形良好,出峰順序依次為雜質D、雜質E、雜質B、雜質C、雜質F、雜質A、2-氨基-4,6-二氯苯酚鹽酸鹽、3,5,6-三氯水楊酸、ABZ和OXY,同時空白輔料不干擾測定,表明本方法專屬性好。
2.2 破壞性試驗由圖3可知,羥氯扎胺阿苯達唑復方混懸液在強酸、強堿、光照、氧化的破壞條件下均有降解,主要降解產物是雜質B(阿苯達唑亞砜);在高溫破壞后主要降解產物是雜質B(阿苯達唑亞砜)和雜質A(5-丙基硫烷基-1H-苯并咪唑-2-胺);在強光和氧化破壞中,主要是ABZ發生降解,其降解產物為雜質B。在各種破壞條件下,羥氯扎胺阿苯達唑主峰與各雜質峰均能良好分離,物料守恒均在90.0%~100.0%,分離度大于1.5,表明在本色譜條件下可以有效分離和檢測各強制破壞降解產物,同時為各有關物質的來源做充分判斷依據。
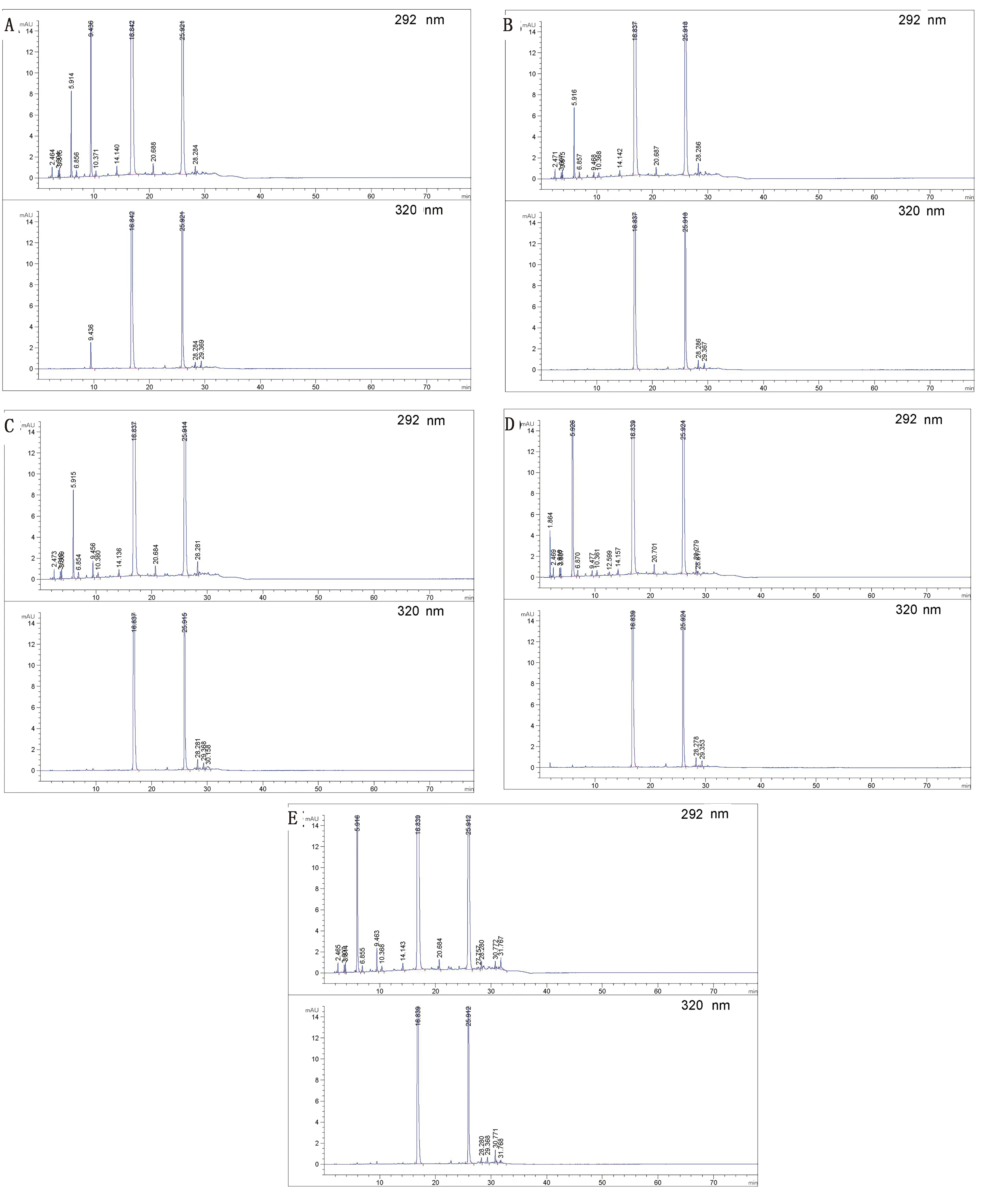
圖3 高溫(A)、酸(B)、堿(C)、氧化(D)和光照(E)破壞的色譜圖
2.3 精密度
2.3.1進樣精密度 連續進樣6次,其各個雜質與主成分的保留時間和峰面積的RSD均小于2.0%,表明儀器精密度良好,儀器較穩定。
2.3.2中間精密度 分別由甲、乙2個人在不同時間,使用不同儀器連續進樣分析6次,其各個雜質與主成分的保留時間和峰面積的RSD均小于2.0%,表明中間精密度良好。
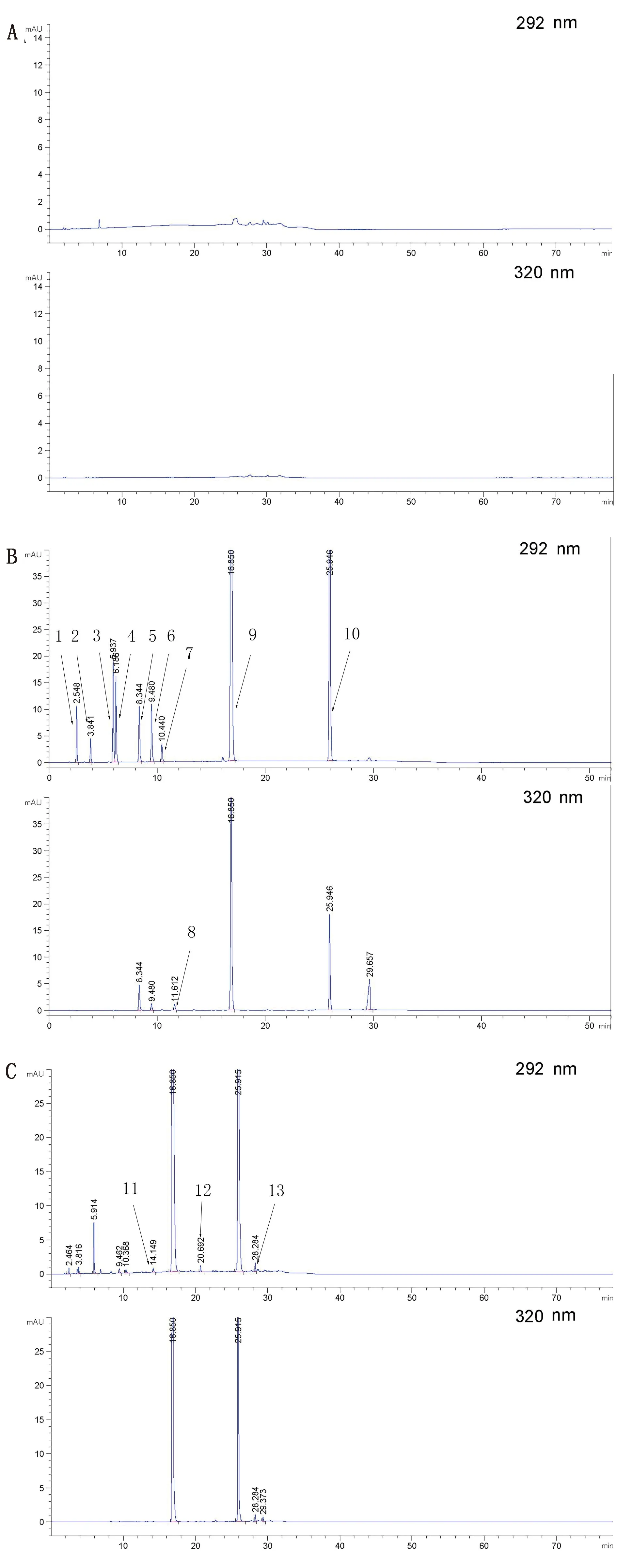
1.雜質D;2.雜質E;3.雜質B;4.雜質C;5.雜質F;6.雜質A;7.2-氨基-4,6-二氯苯酚鹽酸鹽;8.3,5,6-三氯水楊酸;9.ABZ;10.OXY;11.未知雜質1;12.未知雜質2;13.未知雜質3圖2 空白輔料(A)、系統適用性溶液(B)和羥氯扎胺阿苯達唑復方混懸液(C)的色譜圖
2.4 定量限與檢測限試驗結果表明,當S/N為10時,雜質A、雜質B、雜質C、雜質D、雜質E、雜質F、3,5,6-三氯水楊酸、2-氨基-4,6-二氯苯酚鹽酸鹽、OXY、ABZ的定量限分別為0.10,0.06,0.07,0.10,0.15,0.13,5.00,0.30,0.10,0.30 mg/L;當S/N為3時,雜質A、雜質B、雜質C、雜質D、雜質E、雜質F、3,5,6-三氯水楊酸、2-氨基-4,6-二氯苯酚鹽酸鹽、OXY、ABZ的檢測限限分別為0.030,0.020,0.022,0.030,0.050,0.032,2.000,0.100,0.025,0.012 mg/L。
2.5 線性范圍考察以峰面積(A)為縱坐標,質量濃度(ρ,mg/L)為橫坐標進行線性回歸,得到各個雜質與主成分的線性回歸方程,其R2均大于0.999,表明線性關系良好。各個雜質與主成分的線性回歸方程見表2。
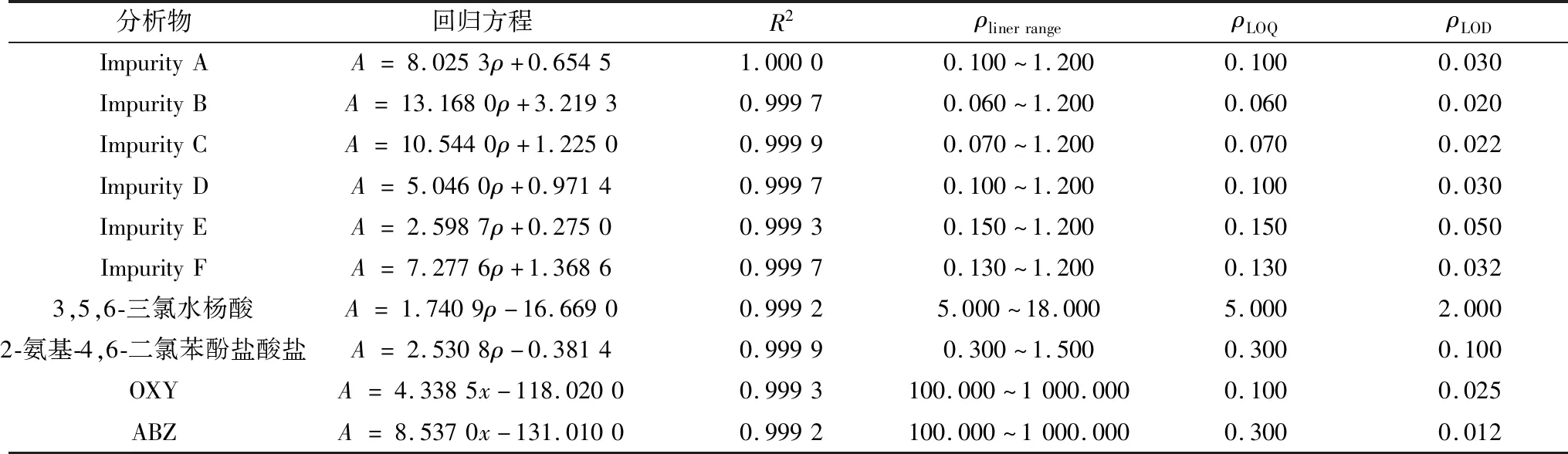
表2 OXY、ABZ及各個雜質的線性、定量限和檢測限結果 mg/L
2.6 重復性試驗結果顯示,主成分OXY、ABZ以及各個雜質的保留時間和峰面積的RSD值均小于2.0%,表明該方法重現性良好,試驗結果見表3。

表3 重復性試驗結果 %
2.7 溶液穩定性在不同時間段對供試品溶液和對照品溶液取樣分析,各個雜質保留時間與峰面積的RSD值均小于2.0%,且無新雜質產生,表明供試品溶液在12 h 內穩定,試驗結果見表4。
表4 溶液穩定性試驗結果 %
2.8 回收率試驗由表5可知,各個雜質及其主成分在低、中、高3個濃度下的回收率均在97.0%~107.0%。
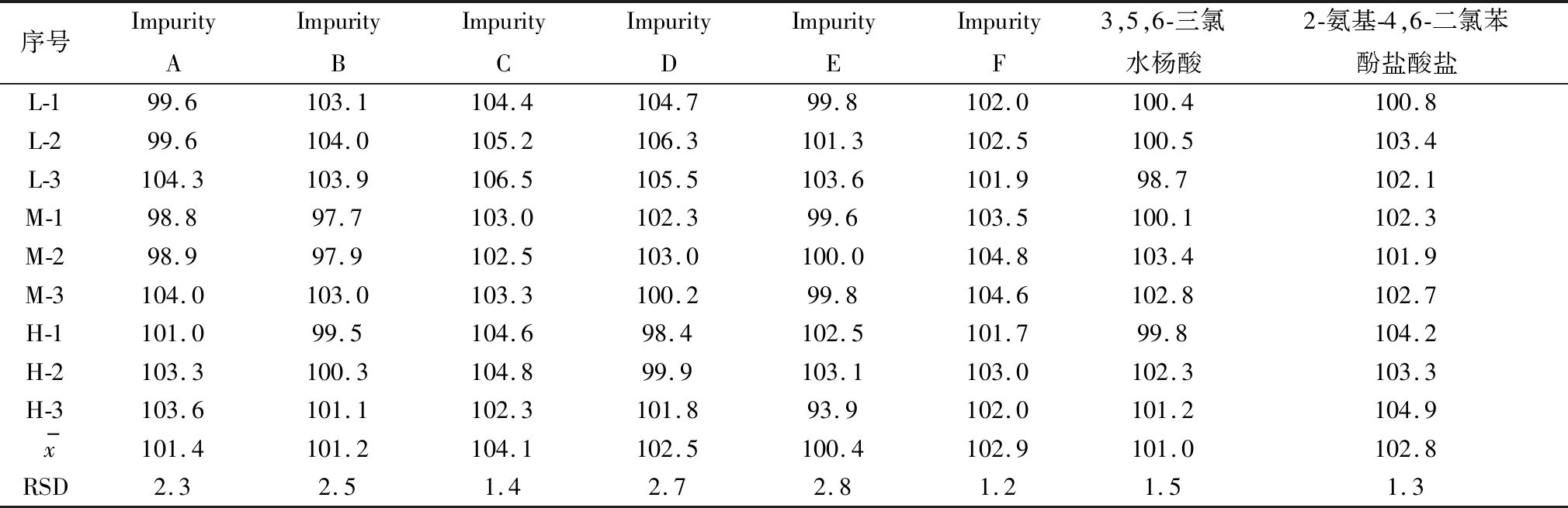
表5 加樣回收率試驗結果(n=9) %
2.9 耐用性試驗結果表明,在各個條件下,主峰與各雜質峰均能分離,且各雜質含量的RSD均小于2.0%,結果說明在流動相比例、柱溫、流速及色譜柱的微小變化對有關物質含量測定結果無顯著性影響,表明該方法耐用性良好。
3.1 有關物質含量測定采用上述色譜方法對3批樣品有關物質進行測定,由表6可知,樣品中含有3個未知雜質,雜質B含量略高但均小于0.5%;所有單雜質含量均小于0.5%,總雜質含量均小于1.0%。
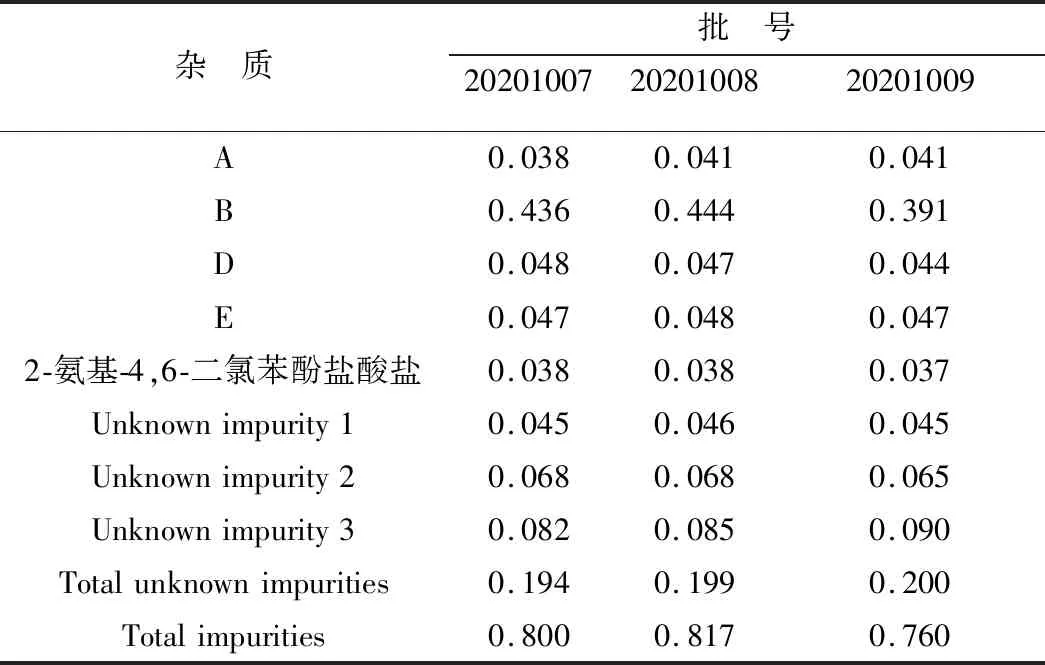
表6 樣品有關物質的測定結果(n=3) %
3 討論
3.1 檢測波長分別取1.4各對照品溶液,用紫外分光光度計進行全波長掃描,雜質3,5,6-三氯水楊酸最大紫外吸收在320 nm處,其余各雜質在292 nm處均有最大吸收,這與英國藥典中ABZ混懸液采用292 nm波長檢測有關物質含量,所采用的波長相一致。同時由于復方制劑各成分之間比較復雜,雜質也相對較多,采用單波長很難充分檢出[15-16],因此,采用292和320 nm雙波長對羥氯扎胺阿苯達唑復方混懸液有關物質進行檢測,靈敏度更高,準確性更好,能夠準確定量檢測。
3.2 色譜條件的確定根據相關文獻[17-19],并參考各化合物的PKa值,本試驗首先對甲醇-水、甲醇-磷酸鹽和乙腈-磷酸鹽3種梯度洗脫系統進行考察,結果發現采用乙腈-磷酸鹽緩沖系統時,2-氨基-4,6-二氯苯酚鹽酸鹽與主成分ABZ分離效果較差,且ABZ和OXY主峰峰形分別出現前傾和拖尾現象。采用甲醇-磷酸鹽緩沖系統時,在OXY主峰后出現未知峰,影響分離度,分別采用鈉鹽、鉀鹽、銨鹽均無法消除。所以本試驗確定以甲醇-甲酸銨緩沖體系作為流動相。其次為改善色譜峰峰形并提高分離度,對流動相添加劑如磷酸、三氟乙酸、甲酸等進行考察,結果顯示0.1%甲酸峰形較好,且各雜質與主峰之間的分離效果最佳,故最終流動相選擇甲醇-0.1%甲酸-10 mmol/L甲酸銨緩沖系統。
3.3 有關物質來源分析通常復方制劑在生產的過程中產生的雜質情況比較復雜,且據2013版《英國藥典》《歐洲藥典》8.0版收藏的ABZ原料藥中有A、B、C、D、E、F 6個雜質,OXY原料藥有2個雜質。對強酸、強堿、高溫、氧化、光照等破壞試驗產生的降解產物進行分析,ABZ、OXY在復方制劑中較為穩定,對酸、堿均不敏感,其有關物質的產生主要與ABZ氧化破壞有關,這與黃毅等[14]結果相一致,說明其主要雜質來源是藥物的氧化產物,可能ABZ分子中含有丙硫基,易被氧化為雜質B(阿苯達唑亞砜),阿苯達唑亞砜是主要的降解產物,且阿苯達唑亞砜作為ABZ的活性物質,是體內發揮殺蟲作用的主要成分,其本身不具有毒性,因此雜質B能間接反映樣品的生產工藝、處方以及貯存條件變化情況,對羥氯扎胺阿苯達唑復方混懸液質量控制有著重要意義。在高溫破壞性試驗中表明未知雜質1主要是由高溫產生,且在ABZ原料藥中也檢測到未知雜質1,因此在儲藏過程中要陰涼處保存;白玉彬等[20]研究表明OXY混懸液有2個未知雜質,是OXY原料藥在合成過程中產生的工藝雜質,用1.3色譜條件對OXY混懸液供試品進樣分析,發現未知雜質2和3與OXY混懸液中的2個未知雜質保留時間一致,故該雜質可能來源于OXY原料藥中。因此需要將已知雜質B和未知雜質1,2,3進行重點控制。
3.4 雜質限度的確定經檢測3批樣品中雜質B的含量分別為0.436%,0.444%,0.391%,未知雜質的總量為0.194%,0.199%,0.200%。在2013版《英國藥典》對于ABZ采用HPLC法測定有關物質,規定單個雜質峰面積不得大于對照品溶液主峰面積的1.0%,各雜質峰面積之和不得大于對照品主峰面積的2倍(2.0%),而2015版《中國藥典》對于ABZ有關物質測定依舊采用薄層色譜法;本團隊在前期建立的HPLC檢測OXY有關物質的方法,規定單個雜質含量小于0.5%,總雜質含量小于1.0%。參考2012版《獸藥研究技術指導原則匯編》中“獸用化學藥物雜質研究技術指導原則”當雜質有特殊的藥理活性時,則雜質的限度可以適當放寬,其制劑的雜質質控限度為1.0%[21]。趙亞萍等[22]制定了ABZ原料藥中有關物質檢查限度為單雜不超過1.0%,總雜不超過2.0%。劉曉敏等[23]制定了丁二磺酸腺苷蛋氨酸腸溶片的質量標準,其中規定雜質甲硫腺苷的含量不得大于2.0%,其他已知雜質不得大于0.5%,單個未知雜質含量不得大于0.2%,未知雜質總含量不得大于1.0%。因此最終確定本混懸液雜質B(阿苯達唑亞砜)含量不得超過1.0%,其他已知雜質含量不得大于0.5%,且已知雜質總和不得大于2.0%;單個未知雜質含量不得大于0.5%,未知雜質總和不得大于1.0%。
