(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸衍生的手性咪唑啉酮催化劑合成新方法
2021-12-02 12:30:08黃志誠李臣毅李文升王立新
合成化學 2021年11期
關鍵詞:催化劑
王 毅, 黃志誠, 向 敏, 李臣毅, 張 健, 李文升, 田 芳, 王立新
(1. 中國科學院 成都有機化學研究所,四川 成都 610041; 2. 中國科學院大學,北京 100049)
咪唑啉酮催化劑是由MacMillan小組開發的一種手性仲胺類咪唑啉酮催化劑,常與酸形成相應的鹽來催化反應。這類催化劑能夠通過烯胺與亞胺離子機理催化α,β-不飽和醛酮類底物的1,3-偶極環加成[1]、Diels-Alder反應[2]、不對稱Michael加成[3-4]以及吡咯與吲哚類底物的不對稱傅克烷基化[5]反應;也能通過SOMO機理對反應底物進行活化[6],催化醛酮類底物發生α-位鹵代反應[7-8],轉移氫化反應[9-10],以及醛類底物的α-位烷基化[11]等;還可以通過協同催化機理與金屬離子共同催化醛類底物的α-位芳基化[12],催化烯烴與醛類底物制備吡咯烷類化合物[13]等。綜上所述,咪唑啉酮類催化劑在不對稱催化領域之中被廣泛應用,是一類非常重要的有機小分子催化劑。
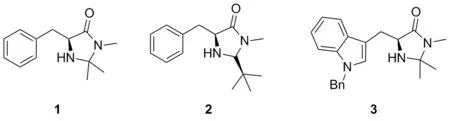
Chart 1
咪唑啉酮類催化劑可以由各類氨基酸衍生得到,常見的幾種咪唑啉酮催化劑如下:
這類催化劑合成的步驟相對簡單,一般先將相應氨基酸酯化,隨后在甲胺的甲醇溶液中反應,得到氨基酸衍生的酰胺產物,最后再與醛酮發生縮合,即得對應咪唑啉酮催化劑[2]。2015年,Gryko課題組[14]嘗試使用了上述方法以(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸為原料合成咪唑啉酮類催化劑,并將其應用于苯丙醛的α-位不對稱光氧化反應之中,最高得到74%的ee值。該課題組以NaCN作為催化劑,氨基酸酯和甲胺反應耗時9 d以96%的收率得到氨基酰胺。3步反應總耗時近10 d,且合成中使用劇毒的NaCN作為催化劑,合成條件苛刻,后處理復雜,反應總收率僅為35%(Scheme1)。
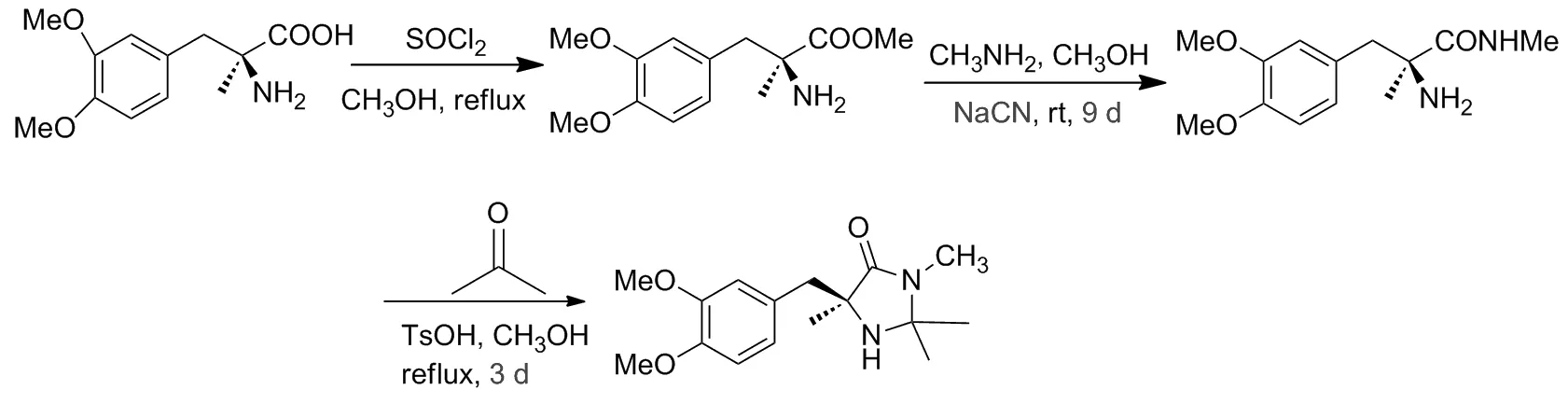
Scheme 1
為改進此催化劑的合成路線,降低合成難度和成本,提出一種合成此催化劑的新方法(Scheme 2),使用相對安全的三光氣代替傳統的劇毒光氣合成N-羧基氨基酸酐(NCA,6)[15],而后與甲胺反應,在室溫下7 h即可定量得到對應(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸甲酰胺7,反應時間大幅度縮短,收率大幅提高,這對于簡化該催化劑的合成、分離及純化具有一定的現實意義。考察了反應溶劑、原料摩爾比和反應溫度等因素對環化反應收率的影響。產物結構經1H NMR和13C NMR確證,與文獻報道一致[14]。
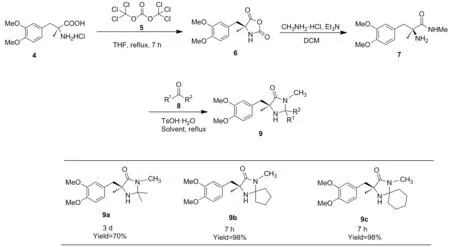
Scheme 2
1 實驗部分
1.1 儀器與試劑
Bruker-300/400 MHz型核磁共振儀(TMS為內標)。
所用試劑均為分析純。
1.2 合成
(1)N-羧基氨基酸酐(6)的合成
稱取(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸甲酰胺0.50 g(1.8 mmol)并將其與15 mL四氫呋喃混合,攪拌使固體懸浮于體系之中。將三光氣1.33 g(4.5 mmol)投入反應體系中攪拌,隨后升溫回流7~8 h。反應體系開始為乳白色懸浮液,隨反應進行逐漸變為均相。冷卻至室溫,減壓除去溶劑,殘余物經過硅膠柱層析[洗脫劑:V(石油醚)/V(乙酸乙酯)=1/1~1/2]純化得純白色固體60.41 g(1.53 mmol),收率85%;1H NMR(400 MHz, CDCl3)δ: 6.80(d,J=8.1 Hz, 1H), 6.70~6.63(m, 2H), 3.85(s, 3H), 3.82(s, 3H), 3.12(d,J=14.0 Hz, 1H), 2.88(d,J=14.1 Hz, 1H), 1.59(s, 3H);13C NMR(101 MHz, CDCl3)δ: 172.3, 151.6, 149.1, 148.8, 125.5, 122.3, 113.1, 111.4, 64.5, 55.9, 55.8, 43.6, 23.8。
(2) 氨基酰胺(7)的合成
稱取N-羧基氨基酸酐0.50 g(1.89 mmol),將其溶解于20 mL二氯甲烷中,冰水浴下向體系中滴加三乙胺0.23 g(2.27 mmol),攪拌15 min后向其中一次性加入甲胺鹽酸鹽0.15 g(2.27 mmol),自然升溫,攪拌約7 h后反應完全(TLC檢測)。過濾除去不溶性的鹽酸鹽,隨后依次用飽和碳酸鈉水溶液(3×10 mL)洗滌,蒸餾水(3×10 mL)洗滌,收集有機相,分液除水后減壓除去溶劑,得淡黃色油狀液體(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸甲酰胺70.47 g(1.85 mmol),收率98%(純度93%,Detected by HPLC);1H NMR(300 MHz, DMSO-d6)δ: 6.82(d,J=8.1 Hz, 1H), 6.71(d,J=1.9 Hz, 1H), 6.64(dd,J=8.1 Hz, 1.9 Hz, 1H), 3.69(d,J=4.6 Hz, 6H), 3.00(d,J=12.9 Hz, 1H), 2.61~2.36(m, 4H), 1.18(s, 3H);13C NMR(75 MHz, CDCl3)δ: 176.9, 148.5, 147.6, 129.4, 122.1, 112.8, 110.6, 58.2, 55.6, 46.3, 27.9, 25.8。
(3) 咪唑啉酮催化劑(9)的合成
9a: 稱取70.47 g(1.85 mmol)溶解于10 mL甲醇之中,充分攪拌溶解后加入5 mL丙酮與TsOH·H2O 0.035 g(0.185 mmol),回流36 h后向體系中補加5 mL丙酮,隨后再回流36 h,減壓除去溶劑,殘余物經過硅膠柱層析[洗脫劑:V(甲醇)/V(乙酸乙酯)=1/10]純化得淡黃色油狀液體9a0.38 g(1.30 mmol),收率70%;1H NMR(300 MHz, DMSO-d6)δ: 6.82(d,J=8.1 Hz, 1H), 6.76(d,J=1.9 Hz, 1H), 6.67(dd,J=8.1 Hz, 2.0 Hz, 1H), 3.68(d,J=2.5 Hz, 6H), 2.86(d,J=13.3 Hz, 1H), 2.58(s, 3H), 2.45(d,J=13.4 Hz, 1H), 1.21(s, 3H), 1.17(s, 3H), 0.75(s, 3H);13C NMR(75 MHz, DMSO-d6)δ: 174.4, 148.2, 147.5, 129.8, 122.4, 114.0, 111.2, 73.9, 62.5, 55.5, 55.4, 44.0, 28.5, 27.4, 27.0, 24.8。
9b: 稱取70.47g(1.85 mmol)溶解于10 mL環己烷之中,充分攪拌溶解后加入2 mL環戊酮與TsOH·H2O 0.035 g(0.185 mmol),回流7 h后(TLC檢測)。反應完畢,減壓除去溶劑,殘余物經過硅膠柱層析[洗脫劑:V(石油醚)/V(乙酸乙酯)=1/1~1/2]純化得淡黃色油狀液體9b0.57 g(1.81 mmol),收率98%;1H NMR(300 MHz, CDCl3)δ: 6.87~6.52(m, 3H), 3.81(d,J=6.5 Hz, 6H), 3.23(d,J=13.9 Hz, 1H), 2.68(s, 3H), 2.54(d,J=13.9 Hz, 1H), 2.25~1.43(m, 8H), 1.35(s, 3H);13C NMR(75 MHz, CDCl3)δ: 175.7, 148.7, 147.8, 128.8, 122.1, 112.7, 110.7, 84.4, 63.4, 55.7, 55.6, 37.4, 36.7, 33.1, 26.2, 25.5, 23.9, 23.6。
9c: 稱取70.47g(1.85 mmol)溶解于10 mL環己烷之中,充分攪拌溶解后加入2 mL環己酮與TsOH·H2O 0.035 g(0.185 mmol),回流7 h后(TLC檢測)。反應完畢,減壓除去溶劑,殘余物經過硅膠柱層析[洗脫劑:V(石油醚)/V(乙酸乙酯)=1/1~1/2]純化得淡黃色油狀液體9c,收率98%;1H NMR(300 MHz, DMSO-d6)δ: 6.81~6.51(m, 3H), 3.61(s, 6H), 2.76(d,J=13.3 Hz, 1H), 2.50(s, 3H), 2.40(d,J=13.4 Hz, 1H), 1.68~0.79(m, 13H);13C NMR(75 MHz, DMSO-d6)δ: 174.6, 148.2, 147.5, 129.9, 122.3, 114.0, 111.3, 75.3, 62.4, 55.5, 55.3, 44.1, 36.2, 35.0, 27.41, 24.9, 24.4, 22.0, 21.9。
(4)N-羧基氨基酸酐(11)的合成
稱取100.50 g(3.0 mmol)并將其與15 mL四氫呋喃混合,攪拌使固體懸浮于體系之中。向體系中加入三光氣2.20 g(7.5 mmol)后持續攪拌,升溫回流2~3 h。反應體系開始為乳白色懸浮液,隨后逐漸成為均相。冷卻至室溫,減壓除去溶劑,冰水浴條件下向體系中加入25 mL正己烷,持續攪拌,隨后有大量固體析出,抽濾得純白色固體110.57 g,不經純化直接投入下步反應中[16]。
(5) 咪唑啉酮催化劑(1)的合成
稱取110.20 g(約1.05 mmol),并將其溶解于5 mL二氯甲烷之中,冰水浴條件下向體系中緩慢滴加30%含量的甲胺醇溶液0.16 g,體系緩慢升至室溫,攪拌過夜。減壓除去溶劑,不經純化即可投入下一步反應之中。向上一步得到的產物中加入5 mL丙酮,5 mL甲醇與TsOH·H2O 0.019 g(0.10 mmol),攪拌至溶解后升溫回流18 h。減壓除去溶劑,殘余物經硅膠柱層析[洗脫劑:V(石油醚)/V(乙酸乙酯)=1/2]純化得無色油狀液體10.18 g(0.84 mmol),收率80%;1H NMR(300 MHz, CDCl3)δ: 7.36~7.07(m, 5H), 3.72(dd,J=6.9 Hz, 4.5 Hz, 1H), 3.08(dd,J=14.1 Hz, 4.4 Hz, 1H), 2.93(dd,J=14.1 Hz, 6.8 Hz, 1H), 2.68(s, 3H), 1.19(s, 3H), 1.09(s, 3H);13C NMR(75 MHz, CDCl3)δ: 173.4, 137.1, 129.5, 128.6, 126.7, 75.5, 59.2, 37.2, 27.2, 25.3, 25.2。
2 結果與討論
2.1 條件優化
本合成路線共有3步,關鍵步為環化反應。開環反應幾乎能夠定量得到氨基酰胺7產物,縮合反應條件相對固定,為得到更高的收率,對環化反應的條件進行了優化,考察了溶劑,配比,溫度,反應時間等因素對反應收率的影響。
(1) 溶劑對環化反應的影響
表1為溶劑對于反應的收率的影響,反應在醚類溶劑中能得到相對較高的收率(Entries 1~4),其中環醚類溶劑如四氫呋喃和1,4-二氧六環等相對于直鏈醚類要好,綜合而言,反應的最佳溶劑為四氫呋喃
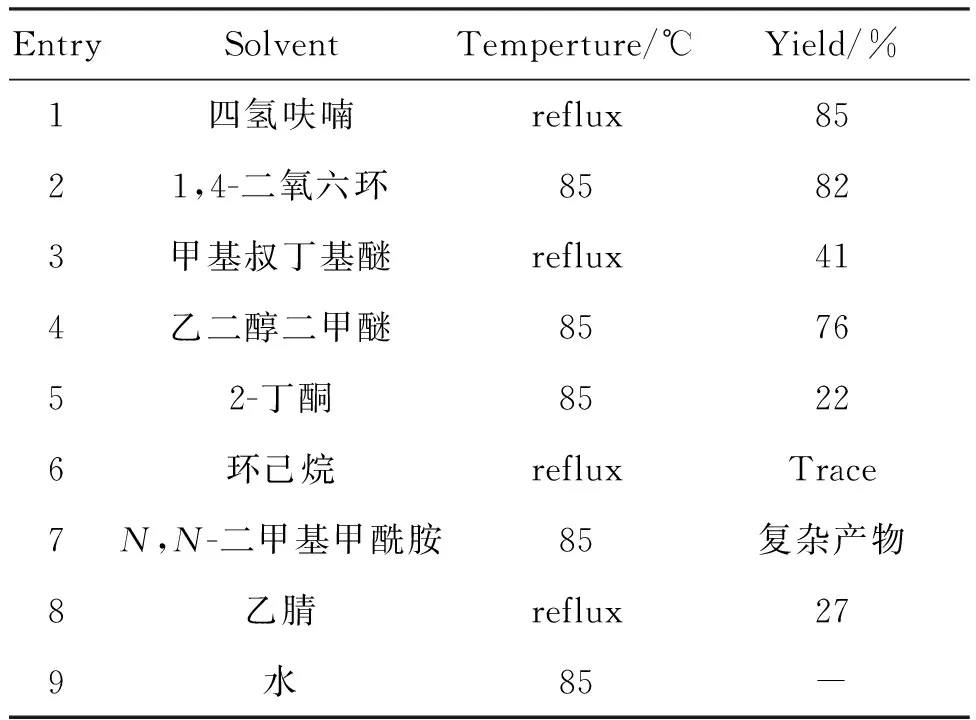
表1 溶劑對反應Ⅰ收率的影響
(2) 原料摩爾比對環化反應收率的影響
在確定最佳溶劑為THF后,決定對原料的比例進行優化,表2為反應原料配比對反應收率的影響,隨著三光氣的比例逐漸增加,6的收率逐漸增加;但是繼續增加三光氣的比例,6的收率并未有進一步提升反而出現下降,因而可以確定反應原料的最佳比例為1/2.5。
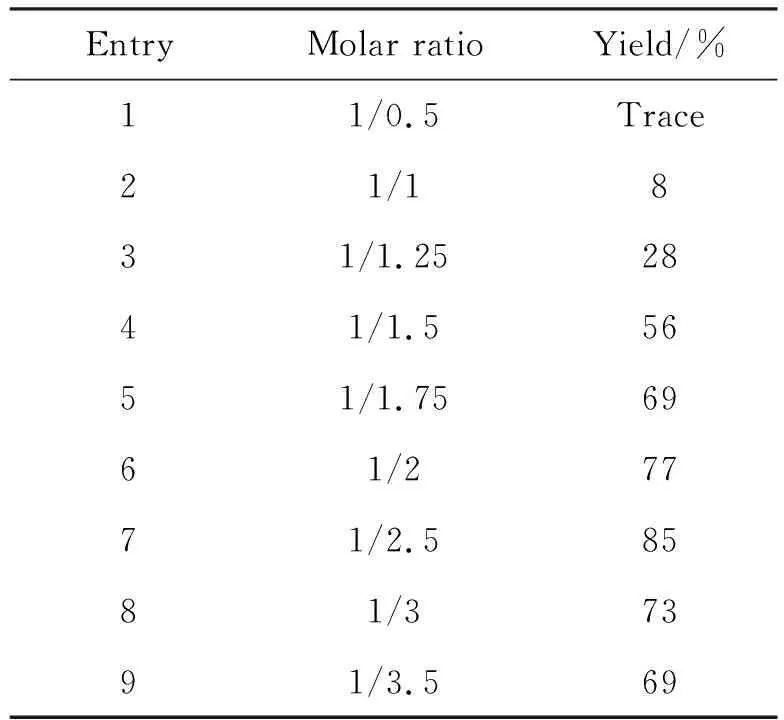
表2 原料配比對反應收率的影響
(3) 反應溫度和反應時間對環化反應收率的影響
在確定最佳溶劑以及最佳反應摩爾比以后,嘗試對反應溫度和反應時間進行了優化,表3為反應溫度和反應時間對環化反應收率的影響。反應收率隨溫度的上升而不斷增加;從中可以看出,在回流溫度下反應7 h收率就能達到85.4%,繼續延長反應時間,收率的增長并不明顯,因而反應最佳溫度即為回流溫度,反應時間為7 h(Entries 4~8)。
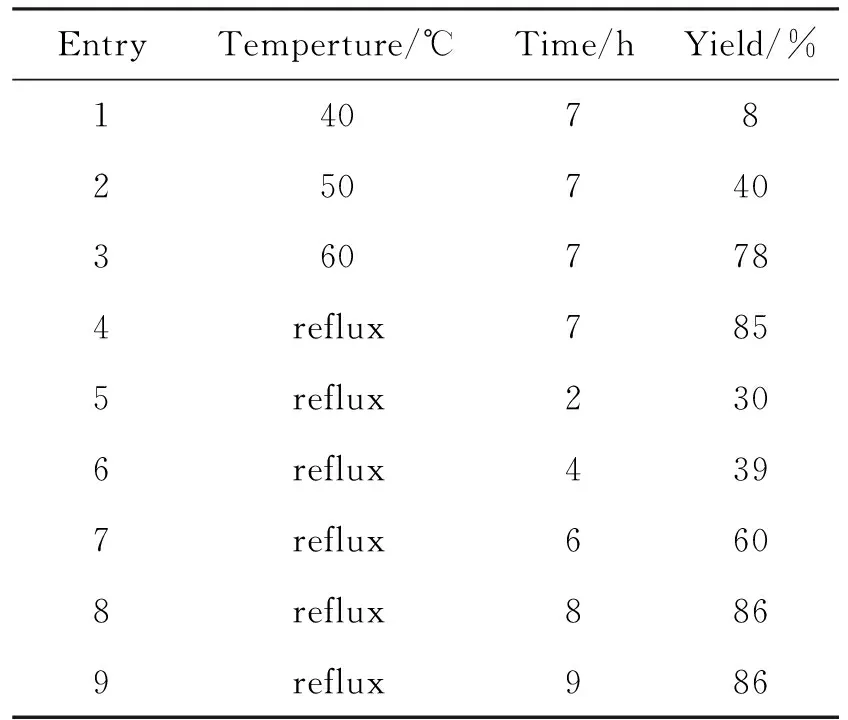
表3 反應溫度和時間對反應收率的影響
綜上所述,環化反應的最佳條件為(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸鹽酸鹽與三光氣比例為1/2.5, THF做溶劑,回流反應7 h。
2.2 反應的底物拓展
(1) 咪唑啉酮類催化劑的合成
將該路線應用于苯丙氨酸制備MacMillan催化劑同樣取得較好的結果,反應總收率能達到80%(Scheme3)。相對于經典合成方法僅59%的總收率[2],本路線收率有很大提高,也證明了本路線可應用于多種咪唑啉酮類催化劑的制備。
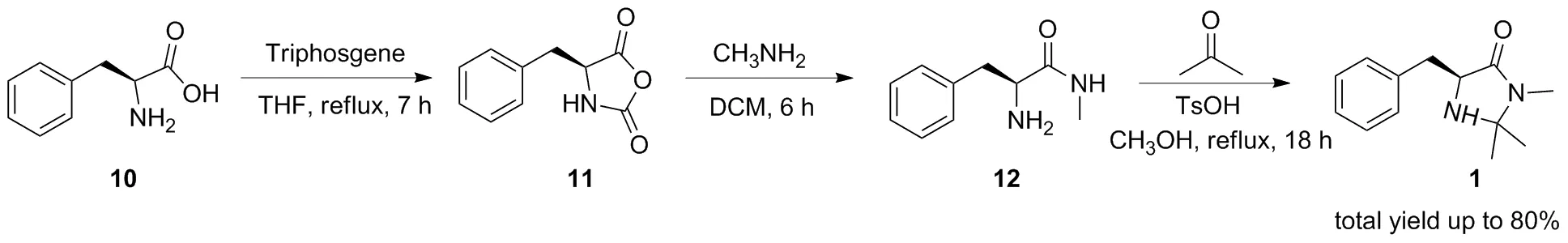
Scheme 3
(2) 縮合反應的底物拓展
對酮類底物進行了拓展,反應均使用10%當量的一水對甲苯磺酸做催化劑。當酮類底物為環戊酮時,反應耗時7 h,能夠以98%收率得到環戊酮衍生咪唑啉酮9b;當酮類底物為環己酮時,反應耗時7 h,能夠以98%收率得到環己酮衍生咪唑啉酮9c。
以(S)-3-(3,4-二甲氧基苯基)-2-甲基丙氨酸鹽酸鹽為原料,設計了一條合成咪唑啉酮類催化劑的新路線,并對其中的關鍵步環化反應進行了條件優化。相對于之前的合成路線,本路線耗時大幅縮短,過程中無需使用劇毒的NaCN催化,合成總收率最高提升至85%;且路線同樣適用于合成其他咪唑啉酮催化劑。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
