甘油磷酰膽堿的分析、制備及純化研究進展
2021-11-30 07:41:38鄒俊康鮑宗必楊啟煒張治國任其龍楊亦文
化工進展 2021年11期
鄒俊康,鮑宗必,楊啟煒,張治國,任其龍,楊亦文
(1 浙江大學化學工程與生物工程學院,生物質化工教育部重點實驗室,浙江 杭州 310027;2 浙江大學衢州研究院,浙江 衢州 324000)
甘油磷酰膽堿的全名為L-α-甘油磷酰膽堿(L-α-glycerylphosphorylcholine,GPC),是人體內卵磷脂分子上的兩條脂肪酸鏈水解后的產物[1]。早在1945 年Schmidt 等[2]便從牛的胰臟中提取出了GPC 粗品,第一次確認了GPC 的結構。由于其結構特性,GPC有助于細胞膜磷脂的合成,從而增強細胞膜的流動性,同時還會對脂質代謝產生積極影響[3]。
GPC能夠穿過血腦屏障,為神經遞質乙酰膽堿的合成提供原料,從而提高海馬體中乙酰膽堿的水平,有效地改善人體膽堿狀態[4],并且通過增加神經細胞的形成、減少神經元的死亡來支持大腦和神經系統的功能[5-6],提升大腦認知能力[7]。此外,GPC能提高基因的表達水平,預防由衰老造成的認知功能下降[8],這也使得GPC 在治療阿爾茲海默癥[9-10]、健忘癥[11]、腦缺血型中風[12]等精神性疾病上有明顯的效果。Strifler等[13]證實GPC是一種靶向線粒體的化合物,能夠減少嚙齒動物體內自由基含量,減輕缺血再灌注引起的損傷和炎癥反應[3,14],具有改善心血管疾病的作用[15]。Szabó等[16]發現GPC能降低輻射誘導的形態學損傷和致死性,顯示出防輻射的作用。Izu 等[17]發現GPC 與S-腺苷蛋氨酸同時使用能夠降低糖尿病模型小鼠的血糖,顯示出治愈糖尿病的可能性。
GPC被認為是無毒且安全的化合物[18],不僅具備治愈各種疾病的潛力,還被廣泛應用于食品、保健品和化妝品等行業,展現出良好的市場前景[19]。但是,國內高純度藥用GPC 幾乎全部依賴進口。為進一步促進GPC 相關產品的研究,本文綜述了最近幾年來GPC 的研究現狀,重點介紹了GPC 的分析、制備和純化技術的研究進展。
1 GPC的分析方法
早在1990 年,意大利就有GPC 上市,波蘭、阿根廷、韓國、俄羅斯等國家也陸續上市了該藥品。但在國內,GPC作為一種三類新藥,還沒有上市。在研究GPC 及其相關產品時,建立全面的分析檢測方法至關重要。趙艷艷[20]采用核磁共振波譜(NMR)、質譜(MS)、紅外光譜(IR)、紫外光譜(UV)等光譜學技術系統地測定了GPC的光譜學數據,確定了GPC 的結構及化學位移歸屬(圖1)。這些傳統方法能定性分析GPC 的存在與否,但無法準確檢測出GPC 的含量。分析檢測得到GPC 濃度和純度對于探究其產品的質量更為重要,因此,下面將從薄層色譜法、核磁共振譜法、滴定法和液相色譜法來綜述GPC的定量分析方法。
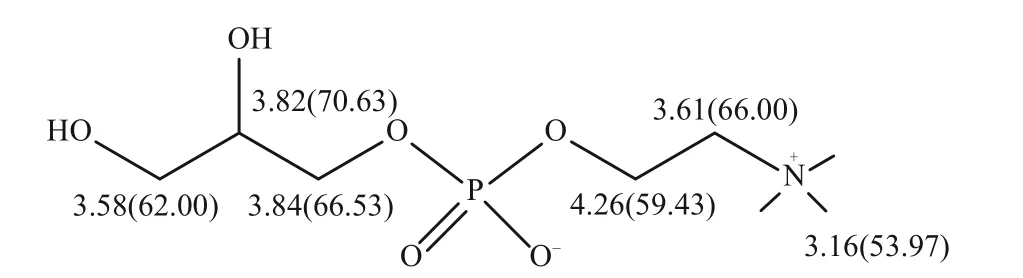
圖1 GPC的化學結構及其化學位移歸屬[20]
1.1 薄層色譜法
薄層色譜由于其操作簡便、快速等特點,通常被用作一種定性檢測方法,用于判斷待測物是否存在。為測定磷酰膽堿酯交換反應所制備的GPC 含量,杜旭佳[21]用硅膠粉和甲基纖維素鈉混合物作為固定相,用5mol/L 的氨水和正丙醇的混合液作為展開劑,按照正丙醇、氨水體積比13∶7配制。經過點樣、展開和染色后使用Adobe Photoshop 軟件對斑點的表面積進行計算,以GPC濃度為橫坐標,斑點面積為縱坐標,建立標準曲線,從而測定GPC的濃度。但此方法對斑點表面積的計算誤差大,使所測得GPC 的濃度不準確,可作為中控方法,判斷反應過程中是否有GPC的生成。
1.2 核磁共振譜法
核磁共振譜法是對物質的結構進行定性分析的強有力工具,Billadello等[22]利用核磁共振磷譜定量分析了組織中的GPC 含量,此方法可以有效測定組織中GPC 的含量,且回收率較高,但此方法檢測結果的變異系數太大,導致所測得含量誤差大。為準確分析大豆卵磷脂水解產物中各雜質的含量,杜章斌等[23]利用核磁共振氫譜法準確定位了GPC、卵磷脂(PC)和溶血磷脂(LPC)的特征質子峰,并根據每個特征性質子峰的相對面積與物質的量的對應關系計算出三種物質的相對含量,此方法可有效避免樣品中水分對檢測結果的干擾,使測定結果更準確。但核磁共振氫譜的圖譜較為復雜,并且測量特征性質子峰的峰面積存在誤差,使得所測量的GPC含量不夠準確。為提高測量的準確率,黎玲玲等[24]成功建立核磁共振磷譜定量法,以磷酸三苯酯為內標,以MEOD-d4作溶劑,以(樣品/內標)的質量比為橫坐標,NMR 峰面積比為縱坐標,建立零截距標準曲線,從而測定藥品中GPC 的絕對含量。該方法簡便快捷、準確可靠,物質鑒定和定量分析可以同步完成。
GPC作為一種手性藥物,其兩種對映異構體的生物利用度、代謝率,代謝產物以及毒性可能存在顯著差異,所以測定GPC 產品的光學純度是至關重要的。De Ferra 等[25]將GPC 的對映異構體與手性硼酸在二甲基亞砜中進行反應(圖2),形成兩種都含有兩個手性中心的非對映體硼酸酯,通過1H NMR 光譜觀察到這兩種非對映體硼酸酯的膽堿甲基和芐甲基所對應的化學位移不同,從而根據相應特征峰面積大小計算出L-α-GPC 對映體的光學純度。核磁共振譜法可用于GPC 的鑒定,也可用于測定最終得到的GPC產品的光學純度。
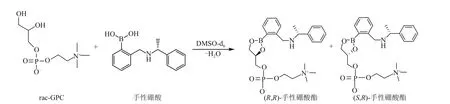
圖2 手性硼酸與外消旋GPC的反應[25]
1.3 滴定法
《美國藥典2019》[26]采用直接滴定法來檢測從PC中提純的GPC,0.1mol/L高氯酸的乙酸溶液作為滴定劑,采用電位法判斷滴定終點,并計算消耗的滴定劑體積,用式(1)計算GPC的質量分數P。
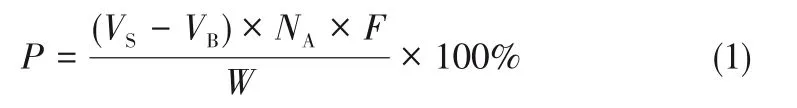
式中,VS為GPC 樣品滴定達到終點時消耗滴定劑的體積,mL;VB為空白滴定終點消耗滴定劑的體積,mL;NA為滴定劑的標準濃度,mEq/mL;F為等效系數,257.2mg/mEq;W為樣品質量,mg。并規定計算值在98%~102%為GPC的合格標準。
劉丹等[27]采用非水電位滴定法,將高氯酸標準溶液作為滴定劑,對GPC 樣品進行滴定,計算滴加滴定劑的體積V和所測量的電壓E,將一系列V、E值進行二級微商處理,用式(2)所示的內插法計算滴定劑的用量。

式中,Vφ為滴定終點時標準溶液的用量,mL;Va為二級微商為a時標準溶液的用量,mL;a為二級微商為零前的二級微商值;b為二級微商為零后的二級微商值;ΔV為二級微商為a至二級微商為b所加標準溶液的體積,mL。
采用二級微商法計算滴定劑的體積更為準確,求出的GPC 含量更為準確,但是此方法在制備GPC樣品時用到了乙酸汞。黃祥元等[28]用乙酸代替乙酸汞溶液加入到GPC 樣品中,這樣既避免使用汞鹽,所得到的滴定突躍現象也更明顯。滴定法測量精度高,常用于提純得到的GPC 產品含量的測定,但是其檢測成本高、操作復雜,樣品中少量的酸性物質也會引起誤差。
1.4 液相色譜法
在定量分析GPC 含量時,高效液相色譜法是最常用的一種方法。劉狄等[29]采用高效液相色譜-蒸發光散射法(HPLC-ELSD),采用硅膠上鍵合1,2-二羥基丙基官能團的Diol 正相硅膠柱為分析柱,甲醇-水為流動相,檢測了不同磷脂中PC 和GPC 的含量。但此方法的流動相中含有極性強的水,長期使用會對Diol正相柱造成損害,降低色譜柱的柱效,對檢測結果有影響。杜章斌等[30]采用Alltech Silica色譜柱,甲醇為流動相,檢測了PC水解液中的GPC 含量,此方法避免了極性強的水對色譜柱的傷害,增強了實驗結果的準確性。
Kielbowicz等[31]開發了一種高效液相色譜-電噴霧檢測器(HPLC-CAD)的檢測方法,采用硅烷基配體官能化的球形硅膠作為固定相,乙腈-甲醇-10mmol/L 乙酸銨溶液作為流動相,此方法有效檢測了PC、LPC 和GPC 的含量,但CAD 檢測器的基線不平穩,雜峰很多,檢測結果誤差大。
為更準確檢測PC 水解產物中GPC 及相關雜質的含量,《美國藥典2019》[26]采用液相-示差折光法(LC-RID)和LC-ELSD 聯用來檢測提純物中雜質的含量。LC-RID 是為了檢測GPC 提取物中甘油的含量,色譜柱為L14 柱(10μm 硅膠化學鍵合強堿性季銨鹽陰離子交換固定相),流動相為乙腈-水,測得甘油和GPC 的相對保留時間分別為0.6 和1.0,并且規定甘油含量小于0.5%為合格。LC-ELSD 法是為了檢測GPC 提取物中的其余有機雜質,流動相按照以下方法配制,并按照表1中的時間進行梯度洗脫,可有效檢測出GPC 提取物中各有機雜質的含量。

表1 流動相比例及其洗脫時間
為檢測化學合成的GPC 及相關雜質的含量,傅超婷等[32]采用二元梯度洗脫程序,選取蒸發光散射檢測器,以甲酸銨的乙腈溶液作為流動相對GPC待測樣品進行第一液相色譜檢測,用陽離子交換色譜柱,檢測出雜質甘油磷酰肌醇(GPI)、氯化膽堿和甘油的含量;以乙腈-水為流動相對GPC待測樣品進行第二液相色譜檢測,用氨基鍵合硅膠色譜柱,檢測出雜質甘油磷酰乙醇胺(GPE)的含量。這種方法能全面有效地檢測GPC 樣品中雜質的含量,能夠更好地控制GPC 產品的質量。但此方法操作較為麻煩,需要切換兩次色譜柱和兩次流動相。陳東英等[33]利用極性硅膠柱作為色譜柱,甲醇-乙腈-緩沖鹽溶液為流動相,采用示差折光檢測器、蒸發光散射檢測器或質譜檢測器,能很好地檢測出磷脂、膽堿、氯化磷酰膽堿(PC-Cl)和GPC的含量,并且各有關物質的分離度均大于1.5,峰形良好。液相色譜法是最常用的定量分析方法,可測定反應液中GPC 及其相關雜質的含量,還可以測定提純后得到的GPC產品的純度。
目前國內沒有GPC 相關藥物、保健品、食品和化妝品的生產,只有生產GPC 原料藥的廠家,所以國內沒有對GPC 的質量規定。國際市場上,用于藥品、食品和保健品中的GPC 都是高純度的GPC,其質量要求可參考美國藥典。化妝品中添加的GPC暫時沒有相關質量標準。
《美國藥典》對GPC 的質量要求如下:①滴定法計算得到GPC 質量分數在98%~102%之間;②乙酸鹽質量分數小于0.1%,氯化物質量分數小于0.02%,硫酸鹽質量分數小于0.02%,磷酸鹽質量分數小于0.005%、甘油質量分數小于0.5%。松醇、β-GPC、蔗糖、絲氨酸、GPE的質量分數都小于0.1%;③0.1g/mL 的GPC 溶液的旋光度在-2.4~-2.8 之間;④細菌總數不超過1000cfu/g,霉菌和酵母菌的總數不超過100cfu/g;⑤8.5g/100mL 或10g/100mL 的GPC 溶 液 的pH 在5.0~7.0 之 間;⑥GPC 固體水分含量小于1.0%,GPC 水溶液的水含量為14%~16%。
對各種分析方法的優缺點進行了比較分析,如表2所示。在反應過程中,可將薄層色譜法作為中控方法快速判斷反應中是否有GPC 生成;進行定量分析時,液相色譜法是最常用的方法,可測定反應液中GPC 的濃度,從而計算反應收率。在分析提純所得GPC 產品的質量時,其含量可用高精度的滴定法來測定,其相關雜質的含量可用液相色譜法測定。但沒有一種檢測器能檢測出GPC 產品相關的全部雜質,對于縮水甘油、氯甘油這類有機雜質,需要RID 檢測器來檢測;對于GPE、GPI 這類雜質,需要ELSD檢測器來檢測。所以,多種檢測器聯用才能有效測定GPC 中相關雜質的含量,進一步判斷GPC的質量是否符合要求。

表2 不同分析方法的比較
2 GPC的制備方法
GPC天然存在于生物物質中,所以早期是通過分離純化技術從生物物質中提取GPC 的。1945 年Schmidt 等[2]便從牛的胰臟中萃取、純化得到了GPC,并確定其結構由膽堿和α-甘油磷酸組成的。但是生物物質中GPC 含量極低,直接提取非常困難,產量很低。目前制備GPC 主要有水解磷脂類化合物和全化學合成兩種方法。
2.1 水解磷脂類化合物
工業上采用水解法制備GPC 的原料為大豆磷脂粉末或蛋黃粉,兩者的主要成分都是PC,PC的sn-1 脂肪酸酰基被水解得到sn-2-溶血磷酰膽堿(sn-2-LPC),再經過酰基自動轉移得到sn-1-溶血磷酰膽堿(sn-1-LPC),最后水解掉剩余的脂肪酸酰基可得到GPC,其反應如圖3。
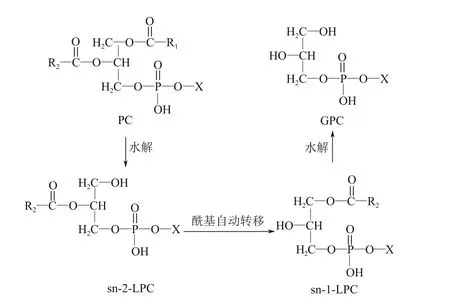
圖3 PC水解的反應過程[34]
2.1.1 化學催化水解法
水解PC 常用的化學催化劑有金屬鈉、醇鈉[35-36],化學催化水解的制備工藝簡單,容易規模化生產,但GPC 的收率很低。PC 在離子交換樹脂[37]的催化下醇解的效果較好,但離子交換樹脂再生困難,并產生大量廢水,不適合工業應用。為解決這些問題,Li等[38]考察了12種低沸點胺類催化劑的催化效果,發現在叔丁胺的催化下,PC 的轉化率可達到98%以上,并且這類低沸點催化劑可以在旋蒸回收甲醇時被同步回收,可實現重復利用。
但是,這些化學催化劑毒性較大,最終得到的GPC 產品中仍然會有少量殘留,使得GPC 產物的安全性存在問題。為了找到更好的催化劑,Li等[39]采用400℃下煅燒的硅酸鈉作為催化劑,在甲醇中催化PC 的水解,PC 最高轉化率能達到99.5%。由于硅酸鈉無毒,易于從人體排泄,所以可以作為生產GPC的良性催化劑。
2.1.2 生物酶水解法
與化學催化劑相比,生物酶的毒性更小,也更安全,具備用量少、催化效率高和易于回收等特點。此方法得到的GPC 適合用于藥物、食品和保健品等口服產品中,是PC水解反應中研究的熱點。
Zhang 等[40]用磷脂酶A1(PLA1)作催化劑并加入輔助因子CaCl2增強PLA1的活性,GPC 的產率能達到94.5%。為進一步提高產率,Zhang 等[41]繼續用Thermo4S-3 作為生物催化劑,并用響應面優化得到最佳反應條件,將GPC 的最大產率提高到96.8%。但是,在水相體系中,PC的溶解度非常有限,導致GPC 的生產效率很低,難以在工業生產中應用。為解決上述問題,Lu 等[34]使用Tween 20作為表面活性劑來改善PC 在水相中的溶解度和分散性,大大縮短了反應時間并提高了GPC的產量。
由于PC 在雙相介質中的溶解度比在水中大得多,Bang 等[42]則考慮用雙相介質來提高PC 的溶解度,以PLA1為生物催化劑在水-己烷的雙相介質中催化PC 水解,使得GPC 的生產效率得到提高。但是,此方法的反應時間為30h,反應進行過于緩慢,不適用于工業生產。Kielbowicz等[31]研究發現,堿性pH 環境可以同時促進酰基遷移和水解過程,為了進一步提高PLA1的催化效率,Li等[43]成功開發一種簡單的共沉淀方法將PLA1封裝在金屬表面活性劑納米復合材料(MSNC)中,并利用堿性的2-甲基咪唑(2-Melm)對這種材料進行改性,得到催化活性增強的2-Melm@PLA1/MSNC 催化劑(圖4)。這種復合材料具有堿性和兩親性,能夠使酰基轉移和酶水解在水-己烷的雙相體系中高效有序地進行,并且具備良好的穩定性,重復利用不會影響其活性。
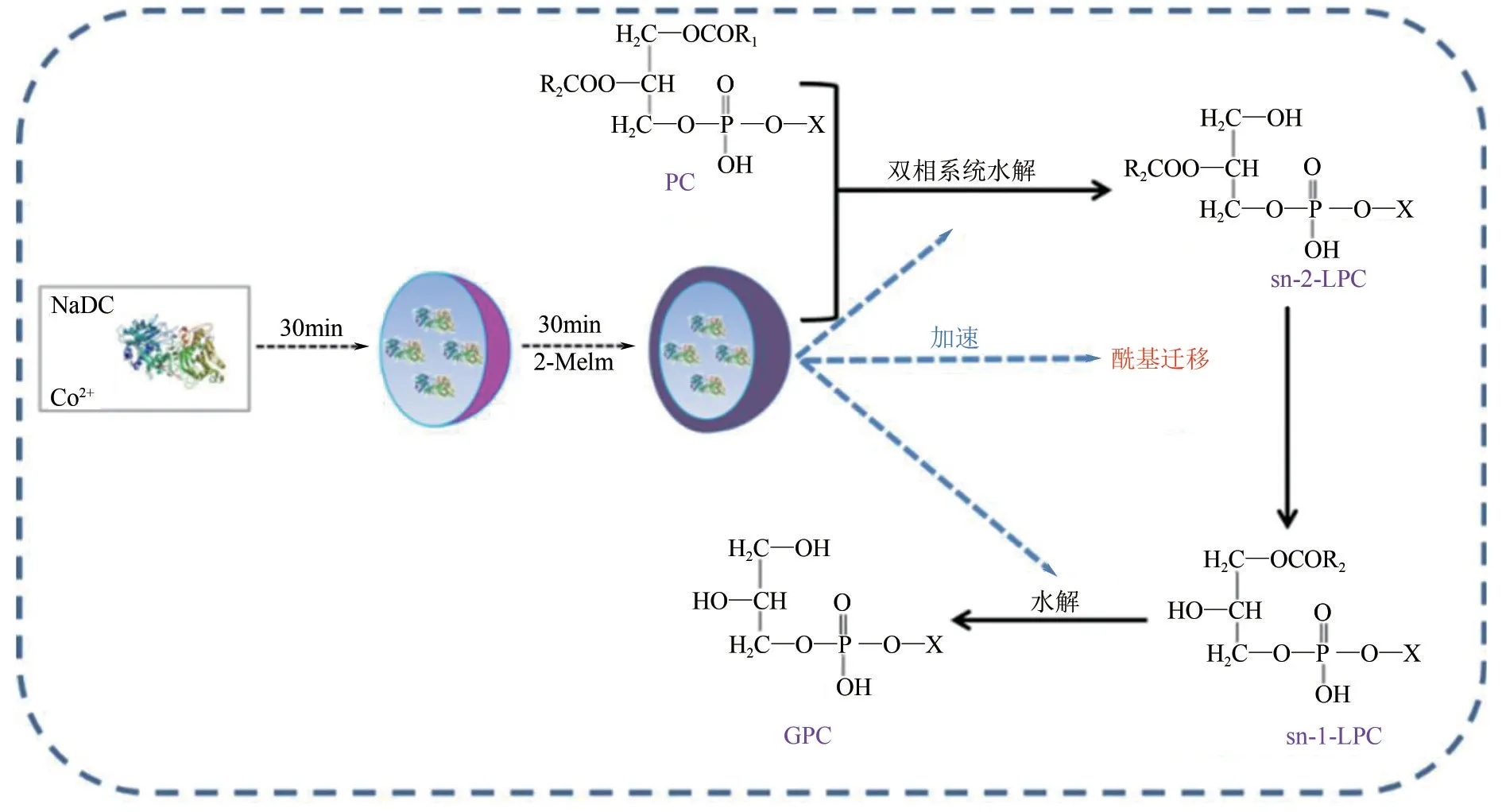
圖4 2-Melm@PLA1/MSNCPC的合成及催化過程[42]
為了使GPC 的品質滿足食品添加劑的要求,Kim 等[44]將大豆卵磷脂置于食品級萃取溶劑正己烷-水的雙相介質中,選擇Novozym435(諾維信脂肪酶435)催化其水解,過濾除去酶后所得濾液,通過雙相介質的簡單相分離即可得到純度為98.6%的食品級GPC水溶液。
2.2 全化學合成法
縮水甘油或氯甘油與磷酰膽堿的鹽反應生成GPC 的收率高(達到90%以上),反應工藝簡單,是目前工業上最主流的合成路線。Park 等[19]以R-縮水甘油為原料,開發了兩種合成GPC 的反應路線。路線1:R-縮水甘油先與苯甲醇進行開環反應,后經乙酰化、氫化,再與甲基苯磺酸膽堿和吡啶反應生成乙酰甘油磷酰膽堿,最后脫去乙酰基即得GPC。路線2:S-縮水甘油與甲基苯磺酸膽堿直接磷酸化得S-縮水甘油磷酰膽堿,再經過開環反應即得GPC。路線2 是在路線1 的基礎上進行的改進,反應路線更短,操作更簡單。
Song 等[45]利用環氧開環的原理,將R-縮水甘油與PC-Cl 在異丙胺的存在下進行親核加成制備GPC,此方法制備工藝簡單,但反應收率偏低,而且反應原料R-縮水甘油極不穩定,保存條件苛刻,增加了生產成本。Lee等[46]在此基礎上,以R-氯甘油為原料,在低溫條件下與NaOH的醇溶液反應生成R-縮水甘油,并立即將所制備的R-縮水甘油與PC-Cl以2∶1的物料比在50~60℃下反應制備GPC,將反應收率提高到70%~92%。Hwang等[47]利用此工藝路線,將450mmol PC-Cl溶解于甲醇中,依次加入900mmol 的KOH 和900mmol 的R-氯甘油,用一鍋法制備出GPC,反應最大收率達到97%。此方法操作簡單,中間產物少,適用于工業生產。PC-Cl的價格較為昂貴,通常不作為工業生產的原料。劉振等[48]則利用磷酰膽堿鈣鹽(PC-Ca)與碳酸鉀水溶液反應制備磷酰膽堿鉀鹽(PC-K),將PC-K 與R-氯甘油反應制備GPC 粗品,此方法也避免用到不穩定的R-縮水甘油,原料的穩定性好且價格低,但是工藝操作比較復雜。尤家棟[49]利用R-氯甘油與NaOH 在乙醇中低溫制備R-縮水甘油,過濾掉析出的鹽后加入PC-Cl高溫回流反應制備GPC,也具備很高的收率。
對各種制備方法的優缺點進行了比較分析,如表3所示。全化學合成法制備GPC具備收率高、產品純度高、制備工藝完善的優點;但起始原料昂貴,最終得到的GPC 產品中也會殘留一些基因毒性雜質,對產品的品質有嚴重影響。利用化學催化水解PC 同樣存在這些問題,化學催化劑是毒性物質會影響產品在食品和藥物中的應用。生物酶通常無毒無害,用生物酶水解PC 是制備食品級GPC 最好的方法,但此方法收率低,產品純度低,對除雜的要求很高,規模化生產難度大。

表3 不同制備方法的比較
3 GPC的純化方法
目前,市場要求GPC 產品的RID 純度達到99.65%,旋光度在-2.4~-2.72,且單一雜質含量不能超過0.1%。但是,化學制備法會產生磷酰膽堿鹽、縮水甘油、氯甘油和甘油等雜質,水解法會產生甘油磷酰乙醇胺(GPE)、甘油磷酰絲氨酸(GPS)、甘油磷酰肌醇(GPI) 等雜質。因此,GPC的純化一直是一個難點。目前常用的純化方法就是溶劑萃取法、結晶法和柱色譜法。
對于化學法制備的GPC粗品,劉振等[48]先用活性炭脫色,再用丙酮萃取除去其中的有機溶劑,最后用D001 大孔強酸性樹脂和711 大孔強堿性樹脂混床柱脫鹽,可得到純度為99.8%的GPC產物。丁建飛[50]采用GPC在無水乙醇中結晶的方法除去GPC中的有機雜質,得到的GPC純度能達到99.5%。于振鵬等[51]考察了醇類、酯類、酮類和乙腈的結晶除雜效果,所得GPC 晶體的純度都達到了99.5%以上。尤家棟[52]將反應液用丙酮萃取,然后用陰性離子交換樹脂柱除去萃取液中的鹽分,最后在無水乙醇中結晶得到精制GPC。
對于水解PC 得到的GPC 粗品,周麗等[53]采用D113 吸附樹脂,將GPE 和鹽類雜質除去,所得GPC純度達到97%。Bang等[42]先將濾液用乙醚萃取三次以除去游離脂肪酸(FFA),接著用硅膠柱除去殘留的FFA 和PC-Cl,從而得到純化的GPC。Zhang 等[54]使用D001 陽離子和D301-111 陰離子交換樹脂柱色譜法成功地將GPC 和GPE 分開,得到的GPC 純度為98.8%。王志強等[55]將水解的PC 反應液直接進行氧化鋁柱層析,再用D001 大孔離子交換樹脂除去鹽分,所得GPC純度可達99%以上。
對各種純化方法的優缺點進行了比較分析,如表4所示。在GPC的純化中,目前最重要的還是結晶法和柱色譜法,結晶法收率低,操作較為復雜,可作為產品純化的最終步驟;柱色譜法除雜效果好,但在除雜過程中會產生大量“三廢”,純化周期長,工業應用成本高。因此,開發適用于工業生產的除雜方法非常重要。

表4 不同純化方法的比較
4 結語
人口老齡化是當今世界發展的一個重要趨勢,老年人的精神健康問題也是全世界研究的熱點,GPC能提高海馬體中的乙酰膽堿水平,支持大腦和神經系統的功能,預防衰老造成的認知功能的下降,在中國這樣的老齡化社會中具備良好的市場前景。
在目前的分析方法中,液相色譜法是GPC 的定量分析最常用的方法,液相色譜配合多種檢測器聯用還能檢測各雜質的含量。薄層色譜法可作為中控方法,判斷反應過程中是否有GPC 的生成。核磁共振譜法可用于GPC 的鑒定,也可用于測定最終得到的GPC 產品的光學純度。滴定法測量精度高,常用于最終得到的GPC產品含量的測定。
在目前的制備方法中,全化學合成法制備GPC具備收率高、產品純度高、制備工藝完善的優點;但起始原料昂貴,最終得到的GPC 產品中也會殘留一些基因毒性雜質,對產品的品質有嚴重影響。利用化學催化水解PC 同樣存在這些問題,這些化學催化劑同樣是毒性物質,也會影響產品在食品上的應用。生物酶通常無毒無害,用生物酶水解PC是制備食品級GPC最好的方法,但此方法收率低,產品純度低,對除雜的要求很高,規模化生產難度大。
在目前的純化方法中,最常用的還是結晶法和柱色譜法:結晶法收率低,操作也較為復雜,可作為產品純化的最終步驟;柱色譜法除雜效果好,但在除雜過程中會產生大量“三廢”,純化周期長,工業應用成本高。
在社會老齡化的背景下,GPC具有良好的市場前景。開發能規模化生產GPC 的工藝路線,適用于工業生產的除雜方法是其關鍵和難點。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
