鎂負載CaO基吸附劑捕集CO2性能及抗燒結機理
2021-11-30 07:42:26孫榮岳彭超陳宇皇朱洪亮
化工進展 2021年11期
孫榮岳,彭超,陳宇皇,朱洪亮
(南京工程學院能源與動力工程學院,江蘇 南京 211167)
鈣循環技術[1]與化學鏈燃燒技術[2]并稱為第二代CO2捕集技術,由于其吸附劑具有成本低廉且分布廣泛、理論捕集容量大以及反應器工藝成熟等優點,被認為是最有可能實現大規模CO2減排的技術之一。以石灰石為代表的鈣基吸附劑循環捕集CO2性能隨循環次數增加迅速衰減,是限制該技術發展的主要問題[3]。為保證較高的CO2捕集效率,在運行過程中需要不斷補充新鮮鈣基吸附劑,同時排出大量失活吸附劑。一臺350MW 機組滿負荷運行,每天需要補充450t 新鮮石灰石,并排出252t 失活CaO[4]。這不僅增加了運行成本,而且會加劇爐膛的磨損、積灰和腐蝕等,對運行造成不良影響。排出如此大量的失活吸附劑,一方面導致了鈣資源的浪費,另一方面也帶來了如何合理處理及有效回收再利用失活吸附劑的問題。
鈣基吸附劑高溫煅燒過程中發生了燒結,CaO晶粒相互融合長大,包覆在晶粒內部的CaO更不易發生碳酸化反應,導致吸附劑活性降低[5]。合成能有效抗燒結的高活性CaO代替石灰石作為鈣循環技術中的吸附劑,可以有效降低循環反應過程中的吸附劑總量以及補充的新鮮吸附劑耗量,提高CO2捕集效率,降低捕集成本。研究者提出了多種高活性CaO 基吸附劑的制備方法,如添加劑修飾[6]、沉淀碳酸鈣(PCC)[7]、惰性載體負載[8]和有機酸改性[9]等。其中,惰性載體負載法向鈣基吸附劑中摻雜具有更高燒結溫度的金屬氧化物作為骨架支撐材料,可以有效延緩CaO晶粒的熔融長大、抑制鈣基吸附劑燒結,是一種非常具有應用前景的高活性復合鈣基吸附劑的制備方法。常用的金屬氧化物有TiO2[10]、MgO[11]、ZrO2[12]、SiO2[13]、Al2O3[14]等。在 這些常用的惰性載體中,MgO 在自然界中含量最高,分布最為廣泛,以其為骨架支撐制備的復合鈣鎂吸附劑具有良好的抗燒結和循環捕集CO2性能。吸附劑中鈣鎂的分散程度對復合鈣鎂吸附劑循環反應活性起決定性作用,鈣鎂來源及制備方法對鈣鎂分散程度存在顯著影響。綜合比較來看,物理混合[15]和共沉淀法[16]制備得到的吸附劑鈣鎂分散均勻性差,所以循環捕集CO2性能提高幅度相對較小。溶膠凝膠法制備得到的鈣鎂復合吸附劑鈣鎂分散性好,制備得到的吸附劑循環反應活性最強,但是制備工藝比較復雜,吸附劑制備成本也最高[17]。燃燒合成法制備工藝相對簡單,并且得到的吸附劑具有比較高的CO2捕集活性及循環穩定性,是一種比較有工業應用前景的制備方法。目前大部分研究中,普遍采用可溶性鹽類作為鈣鎂來源制備復合吸附劑[18-19]。但是可溶性鈣鎂鹽類在自然界中含量少,造成吸附劑成本較高。因此,如能利用主要成分為CaCO3的天然礦石通過燃燒合成法制備得到高活性鈣鎂復合吸附劑,無疑更具工業應用前景。
本研究提出以不可溶的CaCO3或Ca(OH)2作為鈣源,以甘油作為溶劑及燃料,通過燃燒合成法制備鈣鎂復合吸附劑,研究了復合吸附劑循環捕集CO2性能及微觀結構特性。
1 實驗部分
1.1 實驗樣品
復合吸附劑合成所需要的化學試劑均為從上海泰坦科技有限公司采購的商用試劑,級別為分析純(AR)。采用CaCO3(AR,≥99%)或Ca(OH)2(AR,≥95%) 為 鈣 源, 采 用 Mg(NO3)2·6H2O(AR,≥99%)為鎂源,采用甘油(C3H8O3,AR,≥99%)為溶劑和燃料,通過燃燒合成法制備鈣鎂復合吸附劑,具體過程如下:鈣源與甘油和去離子水的比例關系為0.1mol鈣源+100mL甘油+100mL去離子水;通過量筒分別量取100mL 甘油和100mL 去離子水置于燒杯中;CaCO3、Ca(OH)2和Mg(NO3)2·6H2O 等固體顆粒經過篩分后,取顆粒度≤0.125mm的樣品進行實驗;稱取適量的鈣源和鎂源,加入燒杯;在磁力攪拌器上60℃恒溫攪拌1h 后,將燒杯中混合溶液(或懸濁液)分置于剛玉坩堝中,放入馬弗爐在750℃條件下恒溫煅燒30min 合成得到鈣鎂復合吸附劑,具體制備條件及樣品命名如表1所示。合成溫度此處設置為750℃,是因為本文作者課題組前期研究證明在750℃條件下合成得到的復合吸附劑循環捕集CO2性能最佳[20]。Ca8Mg2-CC 表示以CaCO3為鈣源制備的Ca/Mg摩爾比為8∶2的鈣鎂復合吸附劑,Ca7Mg3-CH表示以Ca(OH)2為鈣源制備的Ca/Mg 摩爾比為7∶3 的鈣鎂復合吸附劑,其余依此類推。

表1 鈣鎂復合吸附劑制備條件
1.2 循環煅燒/碳酸化實驗
利用自制的雙固定床反應器研究了制備的鈣鎂復合吸附劑循環捕集CO2性能。自制雙固定床反應器由兩臺管式爐(分別為碳酸化爐和煅燒爐)及相應的配氣機構組成,如圖1所示。鈣循環工藝中為了保證CaCO3能快速完全地分解,煅燒溫度一般取850~900℃[21],此處選擇850℃純N2氣氛煅燒。為了保證循環煅燒過程中CaCO3能完全分解,在循環反應之前進行了預實驗以確定煅燒時間。預實驗結果顯示,無論是未循環的還是多次循環后的復合鈣基吸附劑和分析純CaCO3,經過10min 煅燒后,吸附劑質量基本不再發生變化,表明CaCO3已經完全分解,所以此處煅燒時間取為10min。鈣基吸附劑在600~725℃之間能取得較高的碳酸化速率和轉化率,而受CO2平衡壓力的限制,當碳酸化溫度過高時,鈣循環能取得的CO2捕集效率會下降,因此綜合考慮碳酸化速率和CO2捕集效率,鈣循環工藝一般將碳酸化溫度設為650~700℃[21]。此處選擇碳酸化溫度為700℃,碳酸化氣氛為模擬電廠煙氣中CO2含量,選為15%CO2(N2平衡)。鈣基吸附劑碳酸化一般分為兩個階段:反應快速的化學反應控制階段和反應速度很慢的擴散控制階段[22]。進入擴散控制階段后碳酸化轉化率在幾分鐘的時間內增幅很小,所以碳酸化階段要保證快速反應階段能進行完全。快速反應階段一般持續3~5min,而10min足夠保證快速反應階段進行完全,所以此處碳酸化時間選取為10min。實驗進行前,以30℃/min 的升溫速率將碳酸化爐和煅燒爐分別升至700℃和850℃,然后保持恒溫直至實驗結束,實驗過程中吸附劑在兩個反應器間交替往復實現循環。吸附劑首先在煅燒爐內高溫煅燒,CaCO3或Ca(OH)2完全分解為CaO,煅燒后的吸附劑被送至碳酸化爐內進行碳酸化實驗。利用電子天平(METTLER,ME204)稱量煅燒后和碳酸化后吸附劑樣品質量,通過樣品質量變化計算得到碳酸化轉化率(XN)和CO2吸附量(CN),通過這兩個參數評價其循環捕集CO2性能,計算公式如式(1)和式(2)所示。型號為梅特勒ME204 的電子天平精度為0.1mg,而實驗過程中煅燒后吸附劑樣品質量為300~400mg,每次循環吸收的CO2質量為80~160mg,天平誤差相對較小,不會對結果產生較大影響。為進一步確認,以分析純CaCO3為例,進行了重復性實驗,如圖2所示。發現同一種樣品重復性實驗結果基本一致,可以充分說明天平誤差不會對實驗結果產生較大影響。

圖1 自制雙固定床反應器系統


圖2 分析純CaCO3循環捕集CO2重復性驗證實驗
式中,N為循環反應次數;XN為第N次循環后吸附劑碳酸化轉化率,代表碳酸化生成CaCO3與吸附劑中CaO 的摩爾比;mN為第N次循環后吸附劑質量,mg;mcal為吸附劑煅燒后的質量(吸附劑每次循環煅燒后質量都相同),mg;MCaO和MCO2分別為CaO和CO2的摩爾質量,g/mol;CN為第N次循環后CO2吸附量,代表單位質量煅燒后吸附劑能吸附的CO2質量,g/g;a為初始吸附劑中CaO 質量分數,%,根據制備吸附劑中鈣鎂摩爾比進行計算得到,如式(3)所示,計算結果如表1所示。
式中,r為吸附劑中CaO的摩爾分數,%。
1.3 微觀分析
對初次煅燒和經過20 次循環煅燒后的樣品進行取樣,采用美國FEI Quanta 250 FEG多用途掃描電鏡對不同循環次數煅燒后的吸附劑樣品進行了SEM和EDS分析。
2 結果與討論
2.1 鈣鎂復合吸附劑循環捕集CO2性能
分別以CaCO3或Ca(OH)2為鈣源,選取Ca/Mg摩爾比為8∶2,通過燃燒合成法制備了鈣鎂復合吸附劑,其循環捕集CO2性能如圖3 所示。鈣鎂復合吸附劑循環捕集CO2能力明顯優于CaCO3。第1次循環,Ca8Mg2-CC 和Ca8Mg2-CH 碳酸化轉化率分別為0.84 和0.86,比CaCO3高13.5%和16.2%。隨循環次數增加,CaCO3碳酸化轉化率衰減較快,鈣鎂復合吸附劑表現出更強的循環穩定性。第20 次循環,Ca8Mg2-CC 和Ca8Mg2-CH 碳酸化轉化率仍可達0.48和0.57,比CaCO3高92.0%和128%。制備鈣鎂復合吸附劑過程中,向吸附劑中添加了MgO。MgCO3在450~500℃及以上溫度會發生煅燒反應分解生成MgO 和CO2[23],而此處設定實驗條件中,碳酸化溫度為700℃,煅燒溫度為850℃,均遠高于MgCO3的煅燒分解溫度,MgO 無法與CO2發生碳酸化反應。因此通常在鈣循環捕集CO2過程中,均認為MgO 為惰性成分,不參與循環捕集CO2[24],所以導致了鈣鎂復合吸附劑活性成分減少,會一定程度上降低單位質量吸附劑吸附CO2的量。如圖3(b)所示,第1 次循環Ca8Mg2-CC 和Ca8Mg2-CH 的CO2吸附量略低于CaCO3,但是從第2 次循環開始明顯高于CaCO3。經過20 次循環后,Ca8Mg2-CC 和Ca8Mg2-CH 的CO2吸附量為0.32g/g 和0.38g/g,分別 比CaCO3高60% 和90%。對 比Ca8Mg2-CC 和Ca8Mg2-CH 可以發現,第1 次循環Ca8Mg2-CH 碳酸化轉化率和CO2吸附量略優于Ca8Mg2-CC,20次循環后Ca8Mg2-CH 碳酸化轉化率和CO2吸附量比Ca8Mg2-CC 高18.6%。這說明以Ca(OH)2為鈣源制得的鈣鎂復合吸附劑循環捕集CO2性能優于以CaCO3為鈣源制得的吸附劑。

圖3 鈣鎂復合吸附劑循環捕集CO2性能
2.2 鈣鎂摩爾比對復合吸附劑循環捕集CO2性能的影響
以Ca(OH)2為鈣源,研究了Ca/Mg 摩爾比對復合吸附劑循環捕集CO2性能的影響,結果如圖4 所示。隨著Ca/Mg摩爾比降低,制備鈣鎂復合吸附劑過程中添加Mg的量越多,得到的復合吸附劑中Mg的骨架支撐作用更強,所以循環碳酸化轉化率也越高。如圖4(a)所示,第1次循環Ca7Mg3-CH碳酸化轉化率為0.93,比Ca8Mg2-CH 和Ca9Mg1-CH 分別高7.8%和47.8%。經過20 次循環后,Ca7Mg3-CH碳酸化轉化率仍比Ca8Mg2-CH和Ca9Mg1-CH分別高11.1%和41.6%。當高于7.5∶2.5 后,雖然減小Ca/Mg摩爾比仍能提高復合吸附劑的轉化率,但是提高幅度很小。第1 次和第20 次循環Ca7Mg3-CH碳酸化轉化率僅比Ca7.5Mg2.5-CH高4.3%和3.1%。Ca7Mg3-CH 和Ca8Mg2-CH 碳酸化轉化率隨循環次數增加逐漸降低,而Ca9Mg1-CH 第1 次循環轉化率較低,在初始幾次循環存在自活化的現象。這主要是因為Ca9Mg1-CH中Mg含量較低,在制備過程最后的煅燒階段產生了比較嚴重的預燒結,所以第1次轉化率較低,而在后續幾次循環中有一定程度自活化,然后又隨循環次數增加轉化率降低。
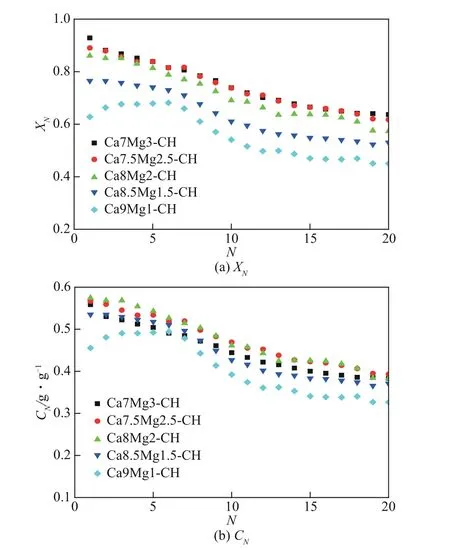
圖4 鈣鎂摩爾比對鈣鎂復合吸附劑循環捕集CO2性能的影響
雖然Mg 添加量越多,復合吸附劑循環碳酸化轉化率越高,但是MgO 在循環反應過程中屬于惰性成分,添加過多的MgO反而會導致活性成分CaO含量降低,吸附劑CO2吸附量下降。如圖4(b)所示,雖然Ca7Mg3-CH比Ca8Mg2-CH循環碳酸化轉化率略高,但是由于其CaO 含量更少,CO2吸附量反而小于Ca8Mg2-CH,并且更多的Mg添加量也意味著吸附劑制備成本會更高。對比圖4(b)可以發現,除前幾次循環稍低以外,Ca/Mg 摩爾比為7.5∶2.5 時復合吸附劑循環反應過程中CO2吸附量與摩爾比為8∶2時相差不大。綜合比較來看,最佳的Ca/Mg摩爾比范圍為(8∶2)~(7.5∶2.5)。
2.3 鈣鎂復合吸附劑長循環捕集CO2性能
以Ca8Mg2-CH 為例,研究了鈣鎂復合吸附劑長循環捕集CO2性能,結果如圖5所示。可以看出,Ca8Mg2-CH 長循環捕集CO2性能明顯優于CaCO3。Ca8Mg2-CH 在前20 次循環捕集CO2性能有一定幅度衰減后趨于穩定,20~50 次循環基本不再衰減。CaCO3前20次循環捕集CO2性能衰減更加劇烈,同時20 次循環后仍有一定幅度衰減。50 次循環后Ca8Mg2-CH 的CO2吸附量為0.37g/g,是CaCO3的2.7 倍。表2 對比了本文制備得到的Ca8Mg2-CH 與其他文獻中報道的鈣鎂復合吸附劑循環捕集CO2性能。本研究制備得到的Ca8Mg2-CH 捕集CO2性能明顯優于羅聰等[15]和Lan 等[24]等通過物理混合方法制備得到的鈣鎂復合吸附劑;比溶膠凝膠法制備得到的鈣鎂復合吸附劑稍差,但勝在制備工藝更加簡單,成本更低[25]。Ca8Mg2-CH 與Yan等[26]通過燃燒合成法制備得到的鈣鎂復合吸附劑相比捕集CO2性能相差不多,比之稍低的原因可能是由于碳酸化時間選取更短所致。綜合來看,本文制備得到的鈣鎂復合吸附劑成本和循環捕集CO2性能方面都具有較強的競爭力,有較強的工程應用前景。

表2 不同方法合成鈣鎂復合吸附劑循環捕集CO2性能對比

圖5 鈣鎂復合吸附劑長循環捕集CO2性能
2.4 微觀結構分析
圖6 和圖7 為不同循環次數煅燒后鈣鎂復合吸附劑和CaCO3的SEM 圖。如圖6(a)所示,第1 次煅燒后的CaCO3晶粒更大、更加密實,這種微觀結構不利于CO2擴散到吸附劑晶粒表面發生碳酸化反應。在燃燒合成過程中,甘油燃燒的同時會伴隨大量的揮發分析出,在吸附劑的表面生成大量的孔隙。如圖6(b)和(c)所示,制備得到的鈣鎂復合吸附劑經過第1次煅燒后,晶粒更加細小,孔隙分布更多,孔與孔之間的連通性更強,與CaCO3相比更有利于碳酸化反應的進行,所以能取得更高的碳酸化轉化率。對比圖6(a)和圖7(a)可以發現,在循環反應過程中CaCO3發生了嚴重的燒結,晶粒融合導致孔隙減少,導致轉化率隨循環次數增加迅速降低。如圖7(b)和(c)所示,鈣鎂復合吸附劑在循環反應過程中燒結較輕,經過20 次循環后保持了較好的孔隙結構。通過氮吸附對第1次和第21次煅燒后鈣鎂復合吸附劑分布在1~80nm 范圍內比孔容和比表面積進行了定量分析,結果如圖8所示。通過燃燒合成制備的鈣鎂復合吸附劑,其煅燒后的比表面積和比孔容均比分析純CaCO3高。第1 次煅燒后Ca8Mg2-CH 和Ca8Mg2-CC 的比孔容分別比分析純CaCO3高33.3%和23.3%,比表面積分別比分析純CaCO3高68.5%和58.5%。MgO 的添加增強了鈣鎂復合吸附劑抗燒結性能,在循環反應過程中孔結構更加穩定,比孔容和比表面積衰減幅度更小。相比于第1次煅燒,第21次煅燒后分析純CaCO3的比孔容和比表面積分別衰減了71.7% 和59.4%。而Ca8Mg2-CH 和Ca8Mg2-CC 經過20 次循環煅燒后,比孔容分別衰減了25.0%和37.8%,比表面積分別衰減了42.9%和44.8%,幅度較分析純CaCO3要小得多。

圖6 鈣鎂復合吸附劑及CaCO3第1次煅燒后SEM分析

圖7 鈣鎂復合吸附劑及CaCO3第21次煅燒后SEM分析
圖9 和圖10 是煅燒后鈣鎂復合吸附劑表面Ca和Mg 元素分布情況,可以發現通過本文方法制備得到的鈣鎂復合吸附劑表面Ca 和Mg 分布均勻。MgO均勻地分布于CaO晶粒之間,有效抑制了循環煅燒過程中CaO晶粒的融合長大,所以鈣鎂復合吸附劑經過多次循環后仍能保持較好的孔隙結構并取得更高的碳酸化轉化率。Ca和Mg的均勻分散程度決定了惰性載體MgO 在抑制CaO 燒結過程中的作用。當用Ca(OH)2作為鈣源制備鈣鎂復合吸附劑時,Ca(OH)2可溶于甘油,Mg(NO3)2可溶于水,而水和甘油又可互溶。因此,Ca(OH)2、Mg(NO3)2、水和甘油混合攪拌后可形成一種均相溶液,在燃燒合成過程中Ca 和Mg 均勻地同時析出,使得Ca8Mg2-CH 中Ca 和Mg 分布均勻,其循環捕集CO2能力也更強。當用CaCO3作為鈣源制備鈣鎂復合吸附劑時,由于CaCO3不可溶于甘油,因此最終CaCO3、Mg(NO3)2、水和甘油混合攪拌后形成一種懸濁液,Mg(NO3)2、水和甘油的溶液經攪拌后進入CaCO3孔隙內部。在燃燒合成的過程中,MgO在CaCO3或CaO內部的孔隙中析出,這就導致了在CaCO3或CaO內部較大孔隙中存在MgO 團聚分布的情況,如圖10(b)所示。所以與Ca8Mg2-CH 相比,Ca8Mg2-CC 中的MgO 分布均勻性更差,其在循環反應過程中的骨架支撐作用也稍弱。圖8中的孔結構參數數據也證明了這一點,在循環反應過程中,Ca8Mg2-CH 的比孔容和比表面積的衰減幅度要小于Ca8Mg2-CC,說明Ca8Mg2-CH的孔結構更加穩定,因此循環捕集CO2活性更優。

圖8 不同循環次數煅燒后鈣鎂復合吸附劑的孔結構參數

圖9 Ca8Mg2-CH的SEM-EDS分析

圖10 Ca8Mg2-CC的SEM-EDS分析
3 結論
(1) 分 別 以CaCO3或Ca(OH)2為 鈣 源,以Mg(NO3)2為鎂源,以甘油為燃料通過燃燒合成法制備得到了高活性鈣鎂復合吸附劑,在循環反應過程中表現出明顯優于CaCO3的循環捕集CO2性能。以Ca(OH)2為鈣源制得的鈣鎂復合吸附劑循環捕集CO2性能優于以CaCO3為鈣源制得的吸附劑,50次循環后CO2吸附量仍可達0.37g/g,具有較強的工程應用前景。
(2)隨著Ca/Mg摩爾比降低,制備鈣鎂復合吸附劑過程中添加Mg 的量越多,得到的復合吸附劑中Mg 的骨架支撐作用更強,碳酸化轉化率越高;但添加Mg的量越多,捕集CO2活性成分含量越少。綜合比較,Ca/Mg 最佳摩爾比為(8∶2)~(7.5∶2.5),此時鈣鎂復合吸附劑CO2吸附量最高。
(3)本文通過燃燒合成法制備得到的鈣鎂復合吸附劑表面Ca 和Mg 分散均勻,MgO 均勻分布在CaO晶粒中間,有效提高了鈣鎂復合吸附劑抗燒結性能及其循環捕集CO2性能。由于在燃燒合成過程中Ca和Mg同時均勻析出,其分布更加均勻,所以以Ca(OH)2為鈣源制備得到的Ca8Mg2-CH孔結構更加穩定,循環捕集CO2性能更佳。
