FeM雙金屬用于甲烷催化裂解制純氫氣和碳納米材料
2021-11-30 07:40:32錢敬俠陳天文劉大斌周呂
化工進展 2021年11期
關鍵詞:催化劑
錢敬俠,陳天文,劉大斌,周呂
(南京理工大學化學與化工學院,江蘇 南京 210094)
近年來,對燃料能源日益增長的需求迫使人們必須尋找除化石燃料以外的可再生能源。現如今,由于大量的廢棄物和COx排放,政府對環境污染物排放有著嚴格的規定,對污染企業有著高額的罰款。因此,開發一種可替代、環境友好型的技術來生產可再生清潔能源至關重要[1]。近幾年,甲烷催化裂解(CDM)制純氫氣和副產物碳納米材料(CNMs)的技術吸引了許多研究者[2-8]。在此吸熱反應中,CDM 技術采用的是簡單的一步裂解過程,如方程(1)所示[9]。

眾所周知,氫氣是一種清潔燃料,廣泛應用于燃料電池、發動機和火箭等設備中[10]。此外,CDM的副產物CNMs可分為碳納米管(CNTs)[11-16]、碳納米洋蔥碳(CNOs)[17-19]和碳納米纖維(CNFs)[11,20-21]等。據報道,CNMs 具有機械強度高、耐強酸強堿、電導率高、比表面積大等優點,有望廣泛應用于鋼鐵和碳基結構材料的制造等領域[10]。因此,CNMs 在市場上的價值可以提高CDM 的經濟可行性。
甲烷由于具有較強的碳氫鍵(440kJ/mol)和分子結構的高對稱性,是最不活躍的碳氫化合物之一。它的裂解只能在高于1200℃的條件下發生,為了降低裂解溫度,金屬基(Ni、Fe、Co、Pd等)[19,22-24]和碳基催化劑(活性炭、炭黑、碳納米管等)[25-26]被用于CDM 反應中。Qian 等[10,27]通過對CDM 反應中催化劑的類型、催化劑的用量、反應器的類型、反應條件、穩定性、活性、甲烷初始和最終轉化率、碳產率、氫產率和產H2成本進行分析對比。結果發現,碳基催化劑的氫產率遠低于Fe 基催化劑,貴金屬和Ni 基催化劑可以提高甲烷的轉化率,但它們價格相對昂貴且稀有。其中,Ni基催化劑的產H2成本是Fe 基催化劑產H2成本的100 倍。因此,Fe 基催化劑用于CDM 過程是一類很有前景的催化劑。
盡管CDM 反應被很多文獻報道為一類產生不含COx的純氫氣技術。但研究發現[11,28],氧化物載體(CeO2、SiO2和Al2O3) 負 載 金 屬 催 化 劑 用 于CDM過程時,微量的COx會被檢測到。雖然氧化物載體可以提高CDM 的催化性能,但為了消除從氧化物載體中產生的“O”,非負載的金屬催化劑用于CDM反應開始被研究[29-34]。近年來,本文作者課題組研究了Fe 基催化劑用于CDM 反應[11,17,35-37],而FeM(M代表金屬)雙金屬催化劑的組成、Fe的用量和催化劑的制備溫度用于CDM 反應中的研究較少,這促使人們進一步研究FeM 雙金屬催化劑用于CDM反應。
本文制備了一系列的FeM(M=Mo、W、Cu)雙金屬催化劑,系統地研究了FeM 雙金屬的甲烷轉化率和碳產率。進一步考察了FexMoy催化劑的不同摩爾比和焙燒溫度對CDM性能的影響,CNMs的形貌也被進一步研究。
1 材料和方法
1.1 材料
Fe(NO3)3·9H2O、 H24Mo7N6O24·4H2O、 H18N2O9W和Cu(NO3)2·3H2O,AR,阿拉丁試劑(上海)有限公司;無水乙醇,AR,上海泰坦科技股份有限公司。
1.2 催化劑的制備
Fe(NO3)3·9H2O、 H24Mo7N6O24·4H2O、 H18N2O9W和Cu(NO3)2·3H2O 分別為Fe、Mo、W 和Cu 的金屬前驅鹽,采用熔融態的方法來制備FexMy雙金屬催化劑,M(M=Mo、W、Cu)為摻雜金屬。例如,FexMy代表Fe 與M 金屬的摩爾比是x∶y。分別取20g 的Fe(NO3)3·9H2O、17.15g 的Fe(NO3)3·9H2O 和0.5g的H24Mo7N6O24·4H2O、15.8g的Fe(NO3)3·9H2O和0.7g 的H18N2O9W、12.5g 的Fe(NO3)3·9H2O 和0.5g 的Cu(NO3)2·3H2O 于瑪瑙研缽中進行研磨,直至將金屬前驅鹽研磨為粉末,分別將研磨好的金屬前驅鹽轉移至不同的坩堝中,混勻;然后將不同的雙金屬催化劑分批于馬弗爐700℃(升溫速率10℃/min)的 條 件 下 焙 燒3h 來 制 備Fe、Fe15Mo1、Fe15W1和Fe15Cu1催化劑。同理,FexMoy雙金屬催化劑的制備過程同上,FexMoy-T代表溫度T下焙燒的FexMoy雙金屬催化劑(本文中未特殊標明催化劑焙燒溫度的,均為700℃)。
1.3 催化劑的表征
N2吸附-脫附曲線由美國麥克公司的ASAP2020 型比表面積和孔隙率分析儀測定,比表面積和吸附-脫附等溫曲線是根據Brunauer-Emmett-Teller (BET) 法 和Barret-Joyner-Halenda(BJH)法計算得到,脫氣溫度300℃,脫氣時間3h。
X 射線衍射(XRD)分析是在德國(Bruker)公司D8 Advance 型X 射線衍射儀上進行的,通過Cu靶Kα輻射,波長λ=0.15406nm,工作電壓40kV,工作電流40mA,掃描范圍2θ為5°~90°。
程序升溫還原(H2-TPR)采用美國麥克公司的AUTO CHEM 2920型測定儀,以10℃/min的升溫速率從室溫程序升溫至300℃干燥預處理,He氣流(50mL/min)吹掃2h,冷卻至50℃,通入10%H2/Ar混合氣(50mL/min)0.5h 待基線穩定后,樣品在50mL/min 的H2/Ar 混合還原氣氛下,以10℃/min 的升溫速率從室溫加熱到900~1000℃,樣品使用量每次為50mg。
采用Thermo 公司型號為Fisher DXR 的顯微拉曼光譜儀(Raman) 表征,測試波長為3000~500cm-1。
樣品的元素組成及化合價態通過X射線光電子能譜儀(XPS,Thermo ESCALAB 250XI)測定。
采用美國FEI公司型號為Tecnai G2 F20的透射電鏡(TEM,加速電壓200kV)測定樣品的微觀結構。
1.4 催化劑的活性測試
甲烷催化裂解反應在固定床反應器中進行,石英管的長度為40cm,內徑為2.1cm。反應溫度由放置在石英管中部的熱電偶控制。催化劑測試之前都未經過純H2的預還原,催化劑被放置在以石英棉為支撐的載體上。反應尾氣經冷凝除水后用氣相色譜儀在線檢測(熱導檢測器,Ar載氣)。
2 結果與討論
2.1 FeM雙金屬催化劑的性能
2.1.1 物理特性
表1 為Fe 和Fe15M1(M=Mo、W、Cu)樣品的比表面積、孔容和孔徑。從表1 可看出,與純Fe(2.7m2/g)相比,Fe15Mo1(11.1m2/g)和Fe15W1(16.4m2/g)的比表面積增大,說明Mo 和W 金屬的摻雜有利于金屬顆粒的分散。Cu 金屬的摻雜可能造成Fe-Cu雙金屬的團聚,使得Fe15Cu1的比表面積減小(0.4m2/g)。

表1 新鮮催化劑的物理特性
2.1.2 XRD
新鮮催化劑Fe 和Fe15M1(M=Mo、W、Cu)的XRD譜圖如圖1所示。圖1中Fe2O3特征衍射峰的位置2θ主要是在24.3°、33.4°、35.8°、40.8°、43.4°、49.8°、54.5°、57.5°、62.3°、64.4°和71.9°處。這些衍射峰的位置與赤鐵礦α-Fe2O3的衍射峰位置相吻合[38]。對于Fe15Mo1催化劑,在2θ為27.3°處有微弱的MoO3特征衍射峰;在22.9°處的衍射峰強度增強,代表Fe2(MoO4)3相的形成。對于Fe15W1催化劑,在2θ為18.6°處有稍強的FeWO4特征衍射峰。對于Fe15Cu1催化劑,在2θ為55.5°處有CuFe2O4相的特征衍射峰;在35.5°處有CuO 的特征衍射峰,此衍射峰與Fe2O3的衍射峰基本重合。結果表明,M 金屬的摻雜改變了Fe2O3的結晶結構,從而影響Fe15M1雙金屬的催化性能。
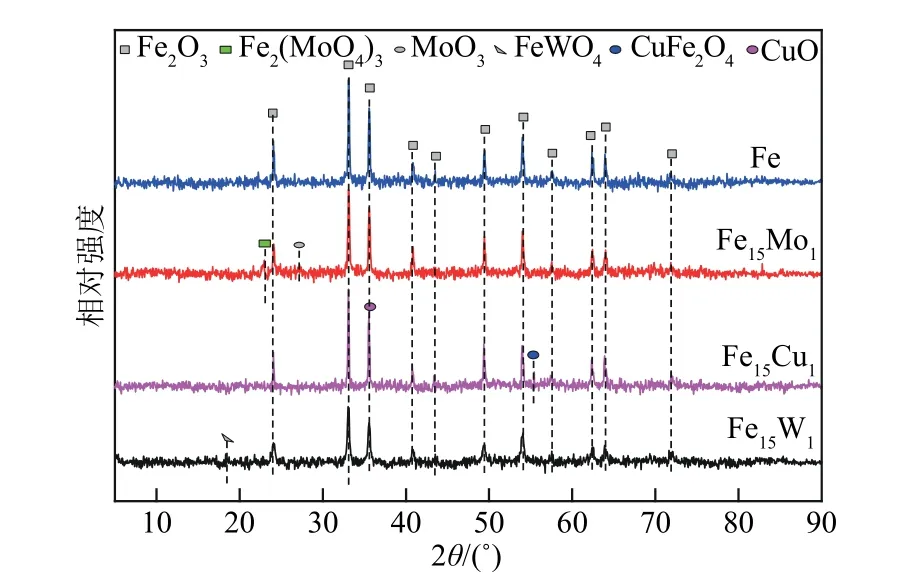
圖1 新鮮催化劑的XRD圖
如圖2 所示,對CDM 反應后的Fe 和Fe15M1的晶體結構進行分析。在2θ為26.5°處有較強的衍射峰,代表著CDM的副產物CNMs具有石墨烯的晶體結構[29]。在2θ為44.7°處的衍射峰是Fe0/Fe3C 的晶相,此外,衍射峰在43.7°代表是Fe3C 的存在[11,39]。綜上所述,反應后的催化劑均表現出Fe3C 和石墨碳的衍射峰,同時也證實了金屬Fe0的存在。

圖2 反應后催化劑的XRD圖
2.1.3 H2-TPR
H2-TPR 的還原曲線如圖3 所示,通過H2-TPR的還原曲線來研究摻雜金屬M 對Fe 金屬催化劑的還原特性和Fe-M 金屬之間相互作用程度的影響。Fe2O3的還原過程為Fe2O3→Fe3O4→FeO→Fe0[11],分析H2-TPR 曲線發現很難區分不同相的還原峰。較低的還原溫度被認為是將Fe2O3還原為Fe3O4[3Fe2O3+H2====2Fe3O4+H2O];同時,Fe3O4可能被進一步還原為FeO[Fe3O4+H2====3FeO+H2O];而部分的FeO 會部分還原為Fe0[FeO+H2====Fe0+H2O]。由圖3可知,與Fe相比,Mo的摻雜使得Fe2O3的還原溫度整體升高,這是由Fe2(MoO4)3相的形成造成的[40]。Cu 的加入可以有效地促進Fe2O3的還原和碳化[41],Fe15Cu1的還原溫度整體降低。W的摻雜使得Fe15W1催化劑的還原溫度整體升高,這是由于Fe-W之間存在更強的相互作用[42]。因而,M金屬的摻雜都不同程度地改變了Fe基催化劑的氧化還原過程。

圖3 新鮮催化劑的H2-TPR曲線
2.1.4 Raman光譜
CDM反應后催化劑的拉曼光譜如圖4所示。在不同的催化劑中,CNMs 的拉曼光譜圖中都觀察到三個分辨良好的拉曼譜帶。第一個拉曼光譜帶位置在1337cm-1(D 帶),第二個拉曼光譜帶位置在1569cm-1(G 帶) 和第三個拉曼光譜帶位置在2683cm-1(2D 帶)。1337cm-1處的D 帶代表非晶態碳或石墨碳的結構缺陷[17]。然而,在圖2的XRD分析中并未檢測到非晶態碳,因此此處的D帶是由石墨碳的缺陷造成的。1569cm-1處的G帶是一種石墨層的平面內碳-碳拉伸振動[34]。2683cm-1處的2D 帶來源于雙原子雙共振[17]。

圖4 反應后催化劑的拉曼光譜圖
由圖4知,Fe15M1(M=Mo、W、Cu)的D帶強度都顯著低于Fe 的D 帶強度,而G 帶強度都略高于Fe 的G 帶強度。金屬M 摻雜到Fe 金屬中使得石墨碳的結構缺陷減小。結晶度和石墨化度可以根據在不同催化劑中沉積的CNMs 的ID/IG值來進一步解釋[34],CDM 反應后的Fe、Fe15Mo1、Fe15Cu1和Fe15W1催化劑的ID/IG值分別是0.95、0.81、0.62 和0.28。結果表明,M金屬的摻雜使得ID/IG值降低,說明有序石墨碳的沉積效果優于缺陷碳的沉積。
2.1.5 催化劑的活性
甲烷轉化率隨時間的變化曲線如圖5所示。在280min內,Fe15M1(M=Mo、Cu、W)的甲烷轉化率均低于Fe 的甲烷轉化率。Fe 的甲烷轉化率最大為33%,300min之后,Fe的甲烷轉化率降至5%。對于Fe15W1催化劑,雖然W的摻雜使得其比表面積增加,但Deck等[43]報道了如果金屬溶碳能力非常低(在反應溫度附近),則不具備催化烴類裂解的能力。因此,W作為摻雜金屬不利于活性組分Fe0/Fe3C在金屬W顆粒表面的富集,不利于催化活性的提高,使得Fe15W1的甲烷轉化率較低(<10%)。Cu金屬的摻雜使得其比表面積減小,減少了甲烷分子與活性位點Fe0/Fe3C 的接觸[11],Fe15Cu1的最大甲烷轉化率為11%。Mo金屬的摻雜使得Fe15Mo1的比表面積增加、孔容增大、孔徑減小,但FexMoy的催化性能與比表面積、孔容、孔徑的相關性較小,見表2。Fe15Mo1的最大甲烷轉化率為28%,在300min 內甲烷轉化率下降至5%左右,說明此催化劑失活很快,穩定性較差。

圖5 FeM催化劑的甲烷轉化率

表2 新鮮催化劑FexMoy的物理特性
對于CDM 過程在Fe 基催化劑上反應機理的研究,已有研究者提出Fe0/Fe3C 作為催化活性位點,副產物CNMs 的沉積是在Fe0/Fe3C 上進行的,隨著CNMs 沉積量的增加,導致催化劑逐漸失活[44-46]。Zhou 等[11]研究了Fe3C 的形成及其在CDM 反應過程中CNMs 沉積的作用。首先,在Fe0活性位點的表面,甲烷裂解為氫氣和無定形炭;當沉積炭超過炭溶解度限制時,Fe3C通過鐵原子重排形成;Fe3C作為CDM 反應的催化劑,CH4將在Fe3C 表面繼續裂解,炭的繼續沉積會導致Fe3C 的過飽和,從而進一步形成Fe3C1+x,而后進一步分解為Fe3C和Fe3C某個結晶區上的石墨碳。
研究者提出通過比較各催化劑最終的碳產率來評價催化性能[47],各催化劑的碳產率如圖6 所示。Fe 的碳產率為4.35gC/gcat,而Fe15Mo1(2.81gC/gcat),Fe15W1(1.35gC/gcat)和Fe15Cu1(0.58gC/gcat)的碳產率均低于Fe 的碳產率。對比圖5 和圖6 可知,Fe、Fe15Mo1和Fe15W1的碳產率與甲烷轉化率曲線相一致,而由于Fe15Cu1在高空速6L/(gcat·h)的條件下失活較快,基本沒有轉化率,因此,采用空速為1.8L/(gcat·h)進行CDM反應。
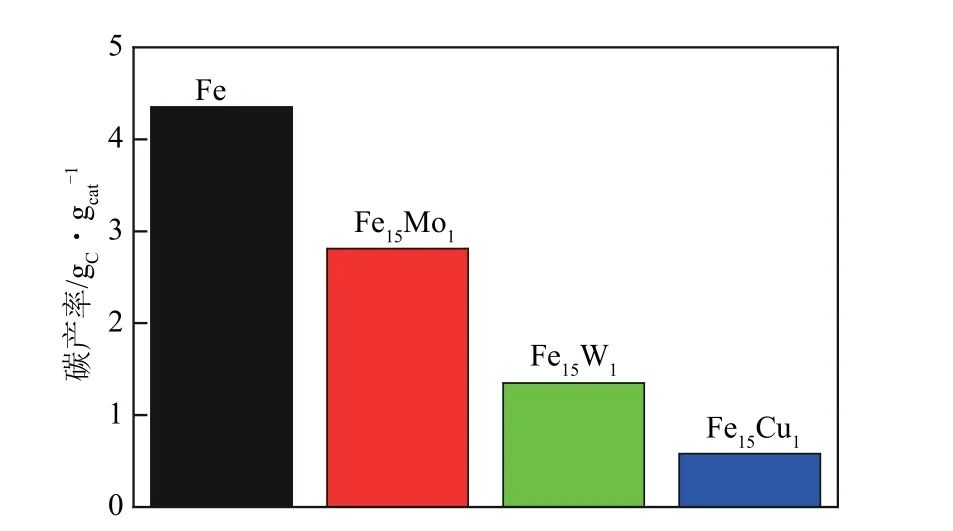
圖6 FeM催化劑的碳產率
已有文獻報道稱Mo[48-49]作為CDM 反應活性金屬(Co、Ni 和Fe)的促進劑,可提高Al2O3和MgO負載的活性金屬的穩定性和分散性。因而,本文進一步探討了無載體負載的條件下,催化劑的焙燒溫度和Mo的添加量對FexMoy催化性能的影響。
2.2 FeMo雙金屬催化劑的性能
2.2.1 物理特性
FexMoy的比表面積、孔容和孔徑如表2 所示。與Fe相比,隨著Mo摻雜量的增加,比表面積和孔容先增大后減小。這是由于Mo 的少量摻雜能夠促進小金屬顆粒的形成[50],提高Fe2O3的分散度[51];Mo的摻雜量逐漸增加,FeMo 雙金屬形成Fe2(MoO4)3相(見圖7),顆粒尺寸變大,比表面積和孔容減小。對比圖13 和圖14 發現,比表面積增加,孔容增大并未使FexMoy的催化活性提高,說明FeMo 雙金屬形成Fe2(MoO4)3相對催化性能的影響較大。
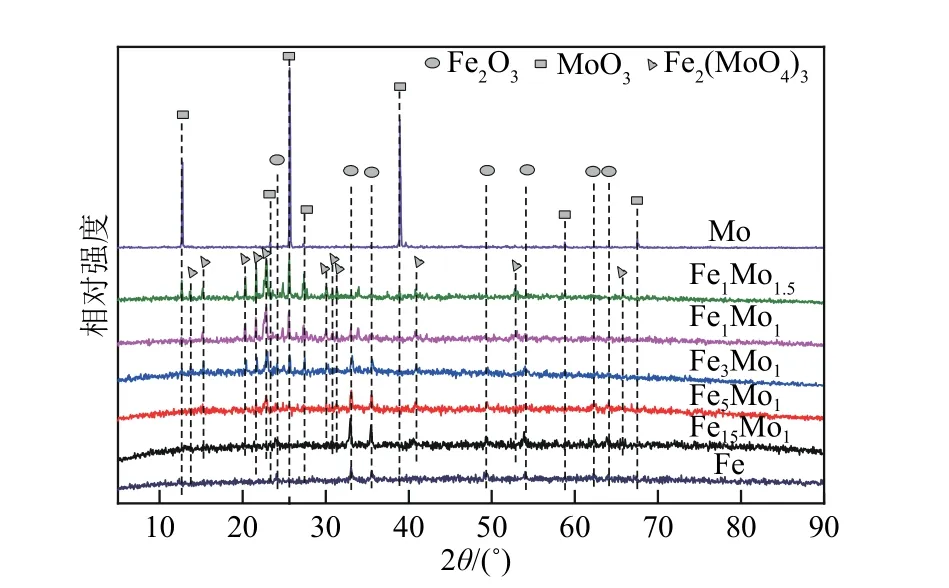
圖7 新鮮催化劑FexMoy的XRD圖
2.2.2 XRD
FexMoy的XRD 圖譜如圖7 所示,在2θ為12.7°、23.3°、25.6°、27.3°、39°、58.6°和67.5°處為MoO3的特征衍射峰。對于FexMoy催化劑,隨著Mo 含量的 增 加,Fe2(MoO4)3相 在13.7°、15.4°、20.3°、21.5°、22.9°、30.1°、30.9°、31.2°、40.8°和52.9°處的衍射峰強度增強。如圖8 所示,對CDM 反應后FexMoy的晶體結構進行XRD 分析。在2θ為26.5°處有較強的衍射峰,代表著CDM的副產物CNMs具有石墨烯的晶體結構[29]。在2θ為44.7°時的衍射峰是Fe0/Fe3C的晶相,此外,衍射峰在43.7°代表Fe3C的存在[11,39]。衍射峰在34.2°、37.7°、39.5°、52.3°、61.8°和69.5°處與Mo2C的衍射峰位置相吻合,代表Mo2C的存在。

圖8 反應后催化劑FexMoy的XRD圖
不同焙燒溫度的Fe15Mo1的XRD 圖譜如圖9 所示,對于Fe15Mo1催化劑,隨著焙燒溫度的增加,樣品在2θ為24.3°、33.4°、35.8°、40.8°、49.8°、54.5°、57.5°、62.3°和64.4°處衍射峰的強度在逐漸增強,說明Fe2O3的結晶度增加。

圖9 不同焙燒溫度的Fe15Mo1的XRD圖
2.2.3 XPS
利用XPS技術對新鮮和反應后催化劑Fe1Mo1表面組分的化學狀態進行研究。圖10(a)是Fe1Mo1的XPS全譜圖,表明催化劑是由C(參考元素)、Fe、Mo 和O 四種元素組成。Fe 2p 的高分辨XPS 能譜[圖10(b)]中,711.2eV和724.3eV處兩個強峰的出現歸 因 于Fe2O3和Fe2(MoO4)3中Fe 2p3/2和Fe 2p1/2的Fe3+[35,52]。Fe 2p3/2峰 在719.3eV 處 有 一 個 伴 隨 衛 星峰,Fe 2p1/2峰在733.0eV 處也有一個伴隨衛星峰。因此,將這兩個伴隨衛星峰都歸屬于Fe3+[53]。Mo 3d的高分辨率XPS 能譜[圖10(c)]中,在結合能為232.7eV 和235.8eV 處的兩個峰歸屬于MoO3和Fe2(MoO4)3結構中Mo 3d5/2和Mo 3d3/2的Mo6+[54]。O 1s的高分辨率XPS 能譜[圖10(d)]中,在529.3eV、530.3eV和530.7eV處的峰歸屬于晶格氧,分別代表Fe2O3、Fe2(MoO4)3和MoO3中 的 氧[55];531.7eV 處 的峰歸屬于表面吸附的氧[55]。這與圖7 所觀察到的Fe1Mo1催化劑中存在Fe2O3、Fe2(MoO4)3和MoO3的特征衍射峰相符。Fe1Mo1的XPS譜圖中每種元素的質量分數分析列于表3。

圖10 新鮮催化劑Fe1Mo1的XPS總譜圖及Fe 2p、Mo 3d、O 1s的高分辨圖譜

表3 Fe1Mo1的XPS譜圖中每種元素的質量分數分析 單位:%
圖11(a)是反應后催化劑Fe1Mo1的XPS 全譜圖,譜圖顯示除了一個較低的O 1s 的峰外,只有一個較強的C 1s 的峰,并未出現Fe 和Mo 元素的峰。圖11(b)為C 1s的高分辨率XPS能譜,在284.9eV處歸屬于CDM 反應后副產物CNMs 的峰。結果表明,CDM 反應后,石墨烯中碳的含量很高,大部分Fe1Mo1被副產物CNMs 包裹。因而,在催化劑的表面并未檢測到Fe和Mo元素。

圖11 反應后催化劑Fe1Mo1的XPS總譜圖及C 1s的高分辨圖譜
2.2.4 TEM
CDM 反應后的Fe1Mo1催化劑的TEM 如圖12 所示。圖12 中CNMs 的結構基本都是竹節狀的CNTs[圖12(a)~(c)]。CDM 的副產物CNMs 的形貌與CNMs在催化劑上的生長機理密切相關。一般情況下,在Fe 催化劑上持續的CDM 反應會使副產物CNMs 析出并在催化劑顆粒表面形成柱狀網絡結晶,最終生長成管狀結構即CNTs。同時,在Fe1Mo1中也可以大量形成一側為Fe3C 的特殊CNTs,稱為竹節狀的CNTs[11]。此外,Lu等[56]推斷催化劑顆粒在一定的時間間隔從石墨鞘中“跳躍”到CNTs 管頂,解釋了在管端具有催化劑顆粒的竹節狀CNTs的形成。

圖12 反應后催化劑Fe1Mo1碳納米管的TEM圖
2.2.5 催化活性的影響因素
如圖13所示,考察了Mo的添加量對甲烷轉化率的影響。隨著Mo 添加量的增加,催化性能逐漸提高。當Mo 的添加量增加至n(Fe)∶n(Mo)=1∶1時,甲烷的轉化率最大為22%且在350min 內下降到17%;當Mo 的添加量繼續增加至n(Fe)∶n(Mo)=1∶1.5 時,甲烷的轉化率最大為24%且在350min內下降到10%,由此可知催化劑的穩定性下降。綜上分析,當n(Fe)∶n(Mo)=1∶1 時,催化劑的活性和穩定性為最佳。如圖14所示,列出了Mo的不同添加量對碳產率的影響。隨著Mo 添加量的增加,碳產率逐漸增加。當n(Fe)∶n(Mo)=1∶1 時,碳產率達到最大6gC/gcat,高于Fe的碳產率4.35gC/gcat。

圖13 Mo的添加量對甲烷轉化率的影響

圖14 FexMoy催化劑的碳產率
Mo 不同比例的摻雜之所以會影響甲烷轉化率和碳產率,是由于隨著Mo摻雜量的增加,FeMo雙金屬形成Fe2(MoO4)3相[見圖7,Fe2(MoO4)3相的形成可提高催化劑的穩定性],而當Mo的摻雜量超過一定值后[n(Fe)∶n(Mo)=1∶1],過量的Mo 形成Mo2C,見圖8。部分Mo 原子取代Fe 原子,催化活性位點Fe0/Fe3C 減少(見圖8),催化活性降低,碳產率減少。
此外,進一步考察了催化劑的焙燒溫度(500~800℃)對催化性能的影響。對于Fe15Mo1,隨著焙燒溫度的升高,Fe-Mo之間的相互作用增強,Fe2O3的結晶度增加(見圖9),結晶度的增加可以有效避免催化劑產生大面積的團聚現象,從而有助于提高其催化活性[57]。因此,如圖15 所示,500℃(1.97gC/gcat)和600℃(2gC/gcat)焙燒的催化劑碳產率均低于700℃焙燒的催化劑碳產率(2.81gC/gcat)。對于Fe1Mo1,隨著焙燒溫度增加至800℃,高溫導致金屬燒結[42],降低了Fe1Mo1-800℃的催化活性。因而,700℃焙燒的催化劑碳產率(6gC/gcat)高于800℃焙燒的催化劑碳產率(5.7gC/gcat)。綜上分析,700℃為最佳焙燒溫度。

圖15 焙燒溫度對碳產率的影響
3 結論
(1)本文采用熔融態的方法制備了一系列的FexMy(M=Mo、Cu、W)雙金屬催化劑,同時,進一步考察了FexMy的催化活性和穩定性。系統地對比了Fe15M1(M=Mo、Cu、W)雙金屬與Fe 的CDM催化性能。結果表明,Fe15Mo1(2.81gC/gcat)、Fe15W1(1.35gC/gcat)和Fe15Cu1(0.58gC/gcat)的碳產率均低于Fe(4.35gC/gcat)的碳產率。
(2)在T=900℃、GHSV=6L/(gcat·h)、100mL/min純甲烷的反應條件下,進一步考察了不同Mo 的摻雜量催化劑(Fe、Fe15Mo1、Fe5Mo1、Fe3Mo1、Fe1Mo1和Fe1Mo1.5)和催化劑的焙燒溫度(500℃、600℃、700℃和800℃)對FexMoy雙金屬的CDM 催化性能的影響。結果發現,700℃焙燒的Fe1Mo1催化劑具有優異的催化活性和穩定性,Fe1Mo1的碳產率(6gC/gcat)高于Fe 的碳產率(4.35gC/gcat)。這是由于Fe1Mo1雙金屬形成Fe2(MoO4)3合金,進一步提高了CDM的催化活性和穩定性。
(3) TEM 結 果表明,以Fe1Mo1為催化 劑,CDM 反應后的副產物CNMs 為竹節狀的CNTs。這為CDM 制純氫氣過程作為一種低成本技術應用于小型或中型的加氫站提供了重要的參考價值。同時,為副產物CNMs創造新的市場機會以尋求基于CNMs行業的誕生提供研究基礎。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
