基于微液節點采樣的定量質譜成像技術在包裝材料中光引發劑定位與檢測中的應用
2021-11-28 05:26:08梁秋菊黃宇軒李優梅王志國
分析測試學報 2021年11期
梁秋菊,吳 倩,黃宇軒,李優梅,王志國,杜 文
(1.湖南中煙工業有限責任公司技術中心,湖南 長沙 410007;2.中南大學 化學化工學院,湖南 長沙 410083)
目前,標準的光引發劑檢測方法主要有氣相色譜-質譜法和液相色譜-質譜法等[4-7]。但這些方法在檢測前需進行紙張破碎、大劑量的溶劑提取以及液液萃取和固相萃取等繁瑣的樣品前處理過程,然后再進行數十分鐘到1 h的色譜-質譜檢測。這些繁瑣的樣品處理過程是質譜分析速度慢、通量小的主要原因。同時,這些過程還會引入更多的檢測誤差和樣品分解,使檢測結果不準確。另外,出于印刷效果的要求,包裝材料的某些重點部位可能會使用絲印或膠印技術,而傳統檢測方法的大面積采樣方式對這些重點部位有稀釋作用,因此有可能出現采用標準方法無法檢出局部超標或超范圍使用光引發劑的可能。
為了提高質譜分析的速度和檢測的準確度,原位質譜分析技術近年來發展快速。常見的原位質譜分析有解析電噴霧質譜(Desorption electrospray ionization,DESI)[8]、實時直接分析質譜(Direct analysis in real time,DART)[9]、激光燒蝕電噴霧電離(Laser ablation electrospray ionization,LAESI)[10]、微液節點采樣技術(Liquid microjunction surface sampling,LMJSS)[11]等。這些技術不但可以原位快速獲得樣品中分析物的相對含量,同時由于采樣探針的空間分辨率,也可得到分析物在樣品表面的空間分布信息,為分析檢測提供更豐富的信息。這些技術各有優勢,其中微液節點采樣技術裝置簡單、成本低,且可原位得到液態樣品,使得其可實現采樣后加標或與其他分離技術聯用,方法靈活多變[12]。但對于固體樣品的原位分析,目前這些方法存在定量困難以及定量結果不具代表性等問題。這是因為這些原位電離方法通常只能對微小的區域進行非消耗性的采樣,使得采樣效率很低且難以預知。同時,由于固體樣品難以進行均勻的加標,因而無法通過標樣實驗得到絕對的采樣效率并進行定量校正。因此,甚少有研究采用這些原位質譜方法進行絕對定量分析。
近期,本課題組針對光引發劑的原位定量分析問題開發了微液節點采樣-質譜檢測方法,并通過動力學校正法有效地解決了光引發劑原位定量的問題[13]。但該方法主要適用于一定面積內光引發劑的原位定量,要將方法應用于光引發劑成像分析還存在空間分辨率低、檢測時間長和操作復雜的問題。本研究在前期工作的基礎上開發了適用于光引發劑定量質譜成像的微液節點采樣-質譜分析方法,將其應用于包裝紙中多種光引發劑的定量成像中,并采用傳統氣相色譜-質譜法分析驗證方法的準確性。本方法可為包裝紙中光引發劑的檢測提供更準確的空間分布信息以及絕對定量信息,為包裝紙中光引發劑的質量監控提供技術支撐。
1 實驗部分
1.1 儀器與試劑
電噴霧離子源-離子阱串聯飛行時間質譜(IT-TOF,日本島津公司);SCION氣相色譜-質譜聯用儀(456-GC-SQ,天美有限公司)。
乙腈、甲酸均為色譜純,購于默克公司(Darmstadt,德國);純凈水購自杭州娃哈哈公司。4-(二甲氨基)苯甲酸乙酯(EDB,99.5%,10287-53-3)、對二甲氨基苯甲酸異辛酯(EHDBA,97.0%,21245-02-3)、苯甲酰甲酸甲酯(MBF,97.0%,15206-55-0)、安息香二乙醚(BDK,100%,24650-42-8)、鄰苯甲酰苯甲酸甲酯(OMBB,100%,606-28-0)、2-羥基-2-甲基-1-苯基丙酮(PI1173, 99.4%, 7473-98-5)、二 苯 甲酮(BP, 100%, 119-61-9)、2/3/4-甲 基二 苯 甲酮(2/3/4-MBP,97%,131-58-8/634-65-2/134-84-9)、1-羥基環己基苯基甲酮(PI 184,99.4%,947-19-3)、2-甲基-1-(4-甲硫基)苯基2-嗎啉基-1-丙酮(PI 907,98.9%,71868-10-5)、2/4-異丙基硫雜蒽酮(2/4-ITX,100%,5495-84-1/83846-86-0)、4-苯基二苯甲酮(PBZ,100%,2128-93-0)、2,4-二乙基硫雜蒽酮(DETX,99.2%,82799-44-8)、4,4'-雙(二甲氨基)二苯甲酮(MK,100%,90-94-8)和4,4'-雙(二乙氨基)二苯酮(DEAB,100%,90-93-7)18種光引發劑單標標樣購于AccuStandard(NewHaven,美國),溶于乙腈配成1 mg/mL混標,于-20℃冰箱中儲存備用。
包裝紙購于超市。
1.2 實驗方法
傳統定量方法:準確裁取主包裝面,面積取10.0 cm×5.0 cm;將裁取的0.5 dm2試樣剪成約0.5 cm×0.5 cm的碎片,進行后續分析。
臺灣應用型本科大學對學生的評價機制靈活多樣,評價主體多元化,評價方法多樣化。不同的課程有相應的評價標準,以實踐課程為例,學生實習單位工作評分占60%,老師教導占10%,實習作業與心得報告占30%。
質譜成像方法:直接取包裝紙的主包裝面,將其固定在樣品臺上直接進行成像分析。
1.2.1 包裝紙中光引發劑的傳統定量檢測根據煙草企業標準方法YQ/T 31-2013《卷煙條與盒包裝紙中光引發劑的測定氣相色譜-質譜聯用法》[14]操作:將剪碎的試樣置于50 mL具塞三角瓶中,加入20 mL水后靜置30 min,再準確加入20 mL乙腈和200μL內標溶液(1 mg/mL氘代蒽),超聲萃取40 min,靜置5 min,取4 mL上層清液于15 mL離心管中,加入3 mL正己烷-乙酸乙酯溶液(體積比3∶7),在渦漩振蕩器上以500 r/min振蕩5 min,靜置,取上層清液凈化。移取(1.5±0.2)mL上層清液于含有150 mg無水硫酸鎂、50 mg PSA和50 mg C18吸附劑的2 mL離心管中,在渦漩振蕩器上以500 r/min振蕩5 min,再以5 000 r/min離心10 min,取上清液供氣相色譜-質譜分析。
氣相色譜-質譜條件:色譜柱為DB-35毛細管柱(30 m(長度)×0.25 mm(內徑)×0.25μm(膜厚),固定相為含5%苯基的甲基聚硅氧烷)。進樣口溫度300℃。載氣為氦氣(純度≥99.999%),恒流流速:1.0 mL/min。進樣量1μL,分流進樣,分流比40∶1。程序升溫:初始溫度70℃,以10℃/min升至300℃,保持5 min,后運行模式在300℃條件下保持5 min。傳輸線溫度300℃;電離方式為電子轟擊源(EI);電離能量70 eV;離子源溫度280℃;四極桿溫度150℃;溶劑延遲6 min。
1.2.2 微液節點采樣-質譜成像離子源:正離子模式,噴霧針電壓4 000 V,離子源溫度200℃,霧化氣(N2)流量1.5 L/min,干燥氣壓力100 kPa。全掃描模式:m/z100~1 000,離子累積時間40 ms;檢測器電壓1.59 kV。不同光引發劑質譜定性的選擇離子質荷比及其對應的離子加合物分別為:EDB(m/z194.117 6,[M+H]+)、EHDBA(m/z278.211 5,[M+H]+)、MBF(m/z165.054 6,[M+H]+)、BDK(m/z257.117 2,[M+H]+)、OMBB(m/z241.085 9,[M+H]+)、PI1173(m/z165.091,[M+H]+)、BP(m/z183.080 4,[M+H]+)、2/3/4-MBP(m/z197.069 1,[M+H]+)、PI184(m/z205.122 3,[M+H+])、PI907(m/z280.136 6,[M+H]+)、2/4-ITX(m/z255.083 8,[M+H]+)、PBZ(m/z259.111 7,[M+H]+)、DETX(m/z269.099 5,[M+H]+)、MK(m/z269.164 8,[M+H]+)和DEAB(m/z325.227 4,[M+H]+)。
微液節點采樣質譜裝置見圖1。微液節點探針由不同直徑毛細管組成的同軸內外套管(外管外徑365μm,內徑250μm;內管外徑150μm,內徑100μm)。外管與PEEK三通的一端相連,而內管穿過三通通過套管和PEEK螺絲在三通的另一端固定。三通的第3個端口與注射泵連接用于給液。根據采樣平臺與質譜的兩種接口,穿過三通一端的內管與質譜有兩種接法(圖1B-1和B-2)。兩種成像方式的采樣溶液均為90%乙腈-9.5%水-0.5%乙酸。成像方式1(圖1B-1):采樣流速調節控制5μL/min,定量環5.5μL,單點采樣時間1 min,采樣完畢后由液相泵用90%乙腈-9.5%水-0.5%乙酸溶液以0.1 mL/min的流速將定量環內的采樣溶液推入質譜;成像方式2(圖1B-2):采樣流速不可調,主要由溶液組成以及質譜離子源噴霧提供的負壓大小決定,本實驗條件下流速穩定在5.5μL/min。采樣溶液直接以此流速流入質譜離子源產生信號。成像方式1和2與質譜聯用的場景照片分別見圖1C-1和1C-2。
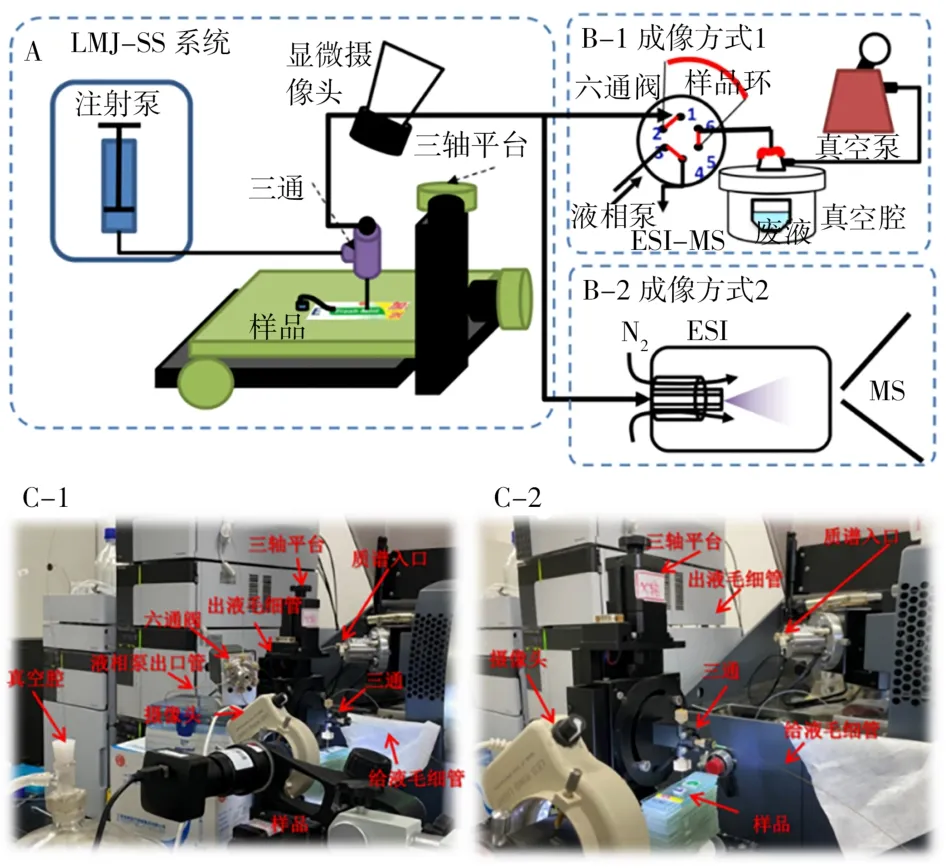
圖1 微液節點采樣質譜成像方式的示意圖(A和B)與實物照片圖(C)Fig.1 Schematic illustrations(A and B)and photos(C)showing the imaging mode of LMJSS-MS
微液節點探針固定在三軸電動平臺上,由顯微攝像頭觀察液節點的形成以及探針-樣品之間的距離以及內外套管的距離,將探針與樣品間的距離以及內外套管的距離均調節為50μm。成像時,由三軸平臺控制探針在樣品表面以200μm/s速度和400μm步間距逐點掃描,每個像素點停留1 min,同時采集每個像素點的質譜信號。
1.2.3 質譜成像的原位定量方法質譜可測得萃取液中分析物信號隨時間的變化情況。分析物信號通過標準曲線(Sa=a*Ca+b)進行定量校正,從而得到分析物的濃度隨時間的變化曲線,然后將每個像素點的濃度-時間曲線進行積分,得到采樣時間內萃取的分析物總量(Mtotal):
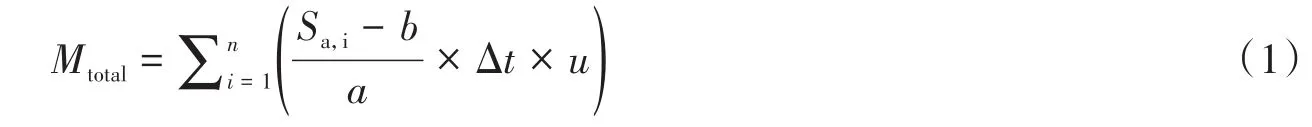
式中Sa,i為單位采樣時間上分析物的信號值,Δt為質譜采樣時間間隔,u為采樣流速。得到Mtotal后便可根據采樣點的直徑(rsampling)計算出分析物在紙張上的單位面積含量(Ctotal,mg/m2):

2 結果與討論
2.1 微液節點采樣以及質譜成像方式的選擇
微液節點采樣是一種基于原位液體萃取的具有空間分辨能力的采樣方法。利用微液節點探針在樣品表面進行掃描采集每個像素點的質譜圖便可得到分析物在樣品表面的質譜成像圖。前期利用微液節點采樣-質譜體系已經建立了包裝紙中光引發劑的原位定量檢測方法[13],該方法通過閥切換的方式實現穩定重復的樣品采集和進樣,雖然在紙張一定面積上掃描的方式可得到最大的檢測靈敏度,但卻降低了方法的空間分辨率,同時質譜成像的連續性也受到切閥過程復雜操作的影響。本研究將該方法改進后用于包裝紙中光引發劑的定量成像。
目前微液節點采樣方法用于質譜成像的接口技術主要有兩種。第1種成像方式如圖1B-1所示:萃取液由注射泵注入同軸套管的外管,而內管先與六通閥1位相連,六通閥6位再與隔膜泵的真空腔相連。通過隔膜泵提供的負壓將液體由內管壓入六通閥2-5位的定量環中進行收集。最后通過閥切換將2-5位的定量環與3-4位相連。由于3、4位分別連接液相泵和質譜離子源,所以閥切換后定量環中收集的樣品被注射進入質譜檢測。這種方式與前期方法[13]的裝置結構一致,只是將探針在一定面積內(約12 mm2)的掃描變為單個采樣點(0.12 mm2)的萃取,以提高采樣的空間分辨率。第2種成像方式如圖1B-2所示,萃取液由注射泵注入同軸套管的外管,而內管與電噴霧噴針相連,通過電噴霧產生的負壓將液體由內管壓入質譜離子源,在探針尖端形成穩定的液節點。
由此可見,成像方式2更加簡單,可實時得到微液節點采樣的質譜信號曲線,對萃取過程進行監測。但其負壓依靠質譜離子源噴霧產生,采樣的溶劑流速難以調節,控制性略差。成像方式1裝置復雜,但其隔膜泵的壓力可調,整個采樣過程可調節的參數更多,穩定性更好,適用范圍更寬。由于兩種方法各有優勢,需要將其進行對比檢測。由于前期研究已獲得光引發劑萃取的最優溶劑為90%乙腈-9.5%水-0.5%乙酸,因此利用該溶劑比較兩種采樣方式對實際包裝紙中光引發劑檢測的靈敏度和重復性(圖2)。由圖可知,成像方式1的采樣信號遠低于成像方式2,對于同一包裝紙成像方式1僅檢出4種光引發劑,而成像方式2可檢出7種光引發劑。其原因主要為:①成像方式1得到的是將萃取液收集到定量環后進樣的平均質譜信號,對萃取液有稀釋作用,無法得到萃取動力學曲線中最高濃度的信號值;②成像方式1收集在定量環中的萃取液需經液相泵中的溶劑推入質譜,這時萃取液又會被稀釋,進一步降低了進樣濃度。由此造成成像方式1的靈敏度低。此外,圖2中也可看出成像方式1的檢測重復性略優于成像方法2,這與其體系的穩定性以及平均了一段時間的采樣信號有關。成像方式2得到萃取分析物信號相對于時間的曲線,其中包含了采樣過程的信號波動和萃取效率波動,綜合兩種方式靈敏度和重復性,選擇靈敏度更高而重復性略差的成像方式2。另外,成像方式2相較于先前開發的原位定量檢測方法[13]在提供同樣空間分辨率的前提下具有10倍以上靈敏度的提高,只是重復性略有降低。
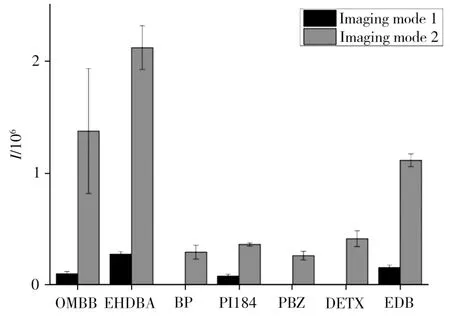
圖2 兩種采樣方式檢測光引發劑的結果對比Fig.2 Comparison of two sampling methods for detection of photoinitiators
2.2 微液節點采樣動力學曲線
成像方式2可得每個像素點采樣的動力學曲線(即信號-時間曲線),由曲線可得微液節點在不同包裝紙位置的采樣速率差別。如圖3所示,選取包裝紙上不同油墨顏色(紅色、綠色和藍色)的幾個位置分別進行微液節點采樣,采樣時間均在1 min以上。由結果可見,不同位置光引發劑信號隨時間的變化都是先上升后下降的過程,這服從異相傳質的動力學理論[15-16]。由前人的推導[15]可知,分析物在兩相間傳質的擴散層一旦建立后,單位時間內分析物的傳質量主要由兩相之間的濃度差以及擴散層厚度決定。由于擴散層厚度在傳質過程中基本不變,所以傳質量主要由濃度差決定。微液節點采樣的萃取液是不斷更新的,所以液相中的分析物濃度可看作實時更新,一直為0,而固相樣品中的分析物濃度則不斷減少,因此,在傳質過程中隨著濃度差不斷減小,單位時間的傳質量也不斷減小,采樣后期分析物信號不斷下降。本課題組在近期研究中推導了單點采樣過程中萃取液中的分析物濃度隨采樣時間的變化曲線[17],由推導公式可知,萃取液中分析物的濃度隨時間呈指數級下降,而下降速度主要由時間常數決定。在模型中時間常數主要受傳質系數和傳質層厚度影響,傳質系數越大、傳質層厚度越薄則時間常數越大,從而下降速度越快。而在峰值之前的信號上升過程主要是擴散層未完全建立,傳質面積不斷擴大產生的,而且這個過程通常時間較短。

圖3 不同包裝紙位置上進行微液節點采樣的信號-時間曲線Fig.3 Signal-time curves of LMJSS at different positions of packing paper
對比3個不同位置的信號-時間曲線(圖3A~C),發現雖然不同位置其曲線上升與下降的速度是不同的,但基本上分析物信號最終都降至0附近。由于最終信號基本降為0,可由1 min內分析物信號隨時間的積分面積除以信號幾近為0的時間內的信號積分面積計算得采樣1 min時的萃取回收率(考慮到在同一樣品點的采樣過程中萃取液中的基質效應基本不變,分析物的離子化效率也基本不變,所以可用信號值代替分析物在萃取液中的濃度值進行計算)。結果顯示,在采樣1 min時,體系對不同包裝紙位置都能完成單個像素點內分析物90%的萃取。由此可知,當單點萃取時間為1 min時,不同位置的萃取效率基本一致,不存在由不同位置萃取效率差別而引起的空間分布以及相對定量檢測的誤差。這種方法無需通過樣品表面加標即可對紙張中的分析物進行絕對定量。因此,后期在進行成像時選擇1 min為每個像素點的采樣時間,通過對采樣時間內的信號-時間曲線進行積分得到每個點的分析物信號值。
對于定量成像而言,除了不同位置萃取效率的差別外,不同位置的基質效應導致的離子抑制或增強同樣會影響定量,通常采用在萃取液中加入內標來消除和校正。而本課題組的研究已證明選取的最優萃取溶劑可采用外標校正法,且基質效應很小[13]。具體操作為將系列濃度的標樣溶于萃取液中利用與實際微液節點采樣同樣的流速和條件進樣于質譜中,標樣的質譜信號用于校正實際信號得到分析物在萃取液中的絕對濃度。由圖3的討論可知,分析物在不同包裝紙位置均能實現90%以上的回收率,所以計算得到萃取液中的待測物濃度可以換算成分析物在一個像素點采樣面積的紙中的含量,用含量除以紙張的采樣面積即可得到分析物在紙中的絕對濃度(計算方法見“1.2.3”)。
2.3 方法在包裝紙中光引發劑成像上的應用
2016年,中國環境保護部科技標準司發布了HJ/T 2524-2016《環境標志產品技術要求膠印油墨》,該標準明確提出能量固化油墨不添加二苯甲酮(BP)、異丙基硫雜蒽酮(ITX)、2-甲基-1-(4-甲硫基苯基)-2-嗎啉基-1-丙酮(907)作為光引發劑[3],EHDBA作為硫雜蒽酮類光引發劑ITX的協同試劑,主要增強ITX在油墨中的固化作用[4],所以ITX和EHDBA的檢測均很重要。2011年,德國宣布召回從比利時進口的冷凍細面條,主要原因是面條包裝上印刷油墨所含有的二苯甲酮(BP)滲入面條中,導致面條被污染,遷移量達1 747μg/kg[18]。由此可見包裝紙中BP檢測以及油墨中光引發劑溯源的重要性。
將建立的微液節點采樣-質譜成像方法應用于包裝紙中多種光引發劑的定量成像(圖4A)。選取涵蓋包裝紙5種顏色的3個區域進行掃描成像,分別是涵蓋紅色、白色、黃色油墨的區域;涵蓋綠色、白色油墨的區域;以及涵蓋藍色、白色油墨的區域。定量成像結果如圖4B的熱圖所示,不同區域能檢測到光引發劑的種類和含量都有很大差別。而在一個區域內,光引發劑的分布也各不相同。將3個區域對比發現,黃紅色區域檢出的光引發劑種類最多,包括PI907、ITX、PBZ、DETX、DEAB、EDB、EHDBA和OMBB,含量在0~5 mg/m2之間,且部分光引發劑的分布與油墨的圖形相吻合。如ITX、PBZ、EHDBA和OMBB 4種光引發劑均集中分布于紅色油墨圖形的區域,而在黃色部分的含量很少。PBZ和EHDBA的分布尤其明顯,只分布在紅色油墨區域,黃色區域幾近于0。特別是EHDBA,其含量較低(約1 mg/m2),分布的集中性會使得傳統大面積裁樣的定量方法難以檢出。本實驗利用質譜成像的空間分辨能力提高了ITX和EHDBA的檢測靈敏度以及空間分布信息,這對于ITX和EHDBA兩種禁用光引發劑的篩查具有重要意義。而綠白色區域檢出的光引發劑與紅黃色區域差別很大,主要包括PBZ、DETX、EDB、EHDBA、OMBB、BP和PI184;光引發劑在綠色和白色位置的分布差別很小,看不出明顯與油墨圖形對應的分布,說明這兩種顏色的油墨采用的光引發劑基本相同。綠白色區域與其他兩個區域相比,BP和PI184的含量(約5 mg/m2)顯著高于其他兩個區域,這為高含量光引發劑的溯源提供了重要信息。在藍白區域,可檢出的光引發劑包括PI907、ITX、PBZ、DETX、DEAB、EDB和EHDBA。這些光引發劑的含量基本與紅黃區域一致,但主要分布在藍色油墨位置,而在白色位置含量很低。
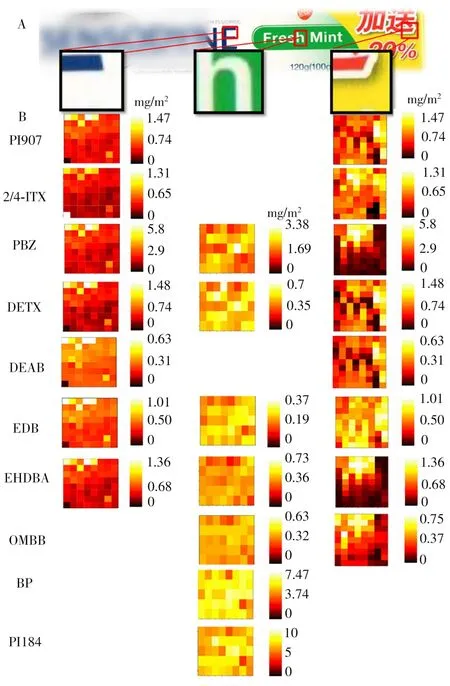
圖4 采用微液節點采樣-質譜系統對包裝紙中不同區域(A)的光引發劑進行成像的定量成像圖(B)Fig.4 Quantitative imaging of PIs in different areas(A)of packaging paper by using LMJSS-MS system(B)
從成像的結果可見,有些光引發劑只在特殊顏色或圖案處才可檢出,這是由于包裝紙通常采用多種印刷工藝制造而成。有些印刷工藝如絲印技術或膠印技術只在包裝紙的很小區域內使用,利用傳統的采樣、前處理和檢測方法進行處理和測試時,僅能得到包裝紙主區域中光引發劑的平均含量信息,可見質譜成像可提供傳統定量方法無法提供的光引發劑空間分辨信息,對光引發劑成分的溯源優勢明顯。
2.4 成像結果與標準質譜定量方法結果的對比
為了驗證成像結果的準確性,將同樣的包裝紙取圖5A所示的紅框位置進行傳統標準方法(氣相色譜-質譜)的定量,前處理及檢測方法見“1.2.1”,結果如圖5B紅柱所示。成像定量結果通過不同顏色區域在標準方法采樣區域對應的面積換算的定量值由圖5B黑柱所示。結果顯示,傳統方法與成像方法的定量結果在一個數量級范圍內,但稍有偏差。如定量結果相差比較大的BP,由于其在使用質譜檢測時信號很低,因此信號-時間曲線中難以判斷其絕對的采樣回收率,同時也造成其檢測誤差較大。另外還有一些光引發劑只能在一種方法中檢出,如傳統方法中檢出的MBP利用成像方法無法檢出,這是因為傳統方法采用氣相色譜-電子轟擊離子源-質譜檢測,而成像方法采用電噴霧離子源質譜檢測,兩種電離方式的電離范圍不同,電噴霧電離源難以檢測MBP這種低極性化合物。而成像方法檢出的3種光引發劑ITX、EDB和EHDBA利用傳統方法無法檢出。由此可見,質譜成像對于空間分布集中的一些光引發劑具有提高靈敏度的作用,傳統定量方法采用大面積采樣的方式,其對分布集中的光引發劑造成很大的稀釋,以至于無法檢出。本文建立的成像方法對這些禁用或限用光引發劑的篩查和檢測具有重要意義。
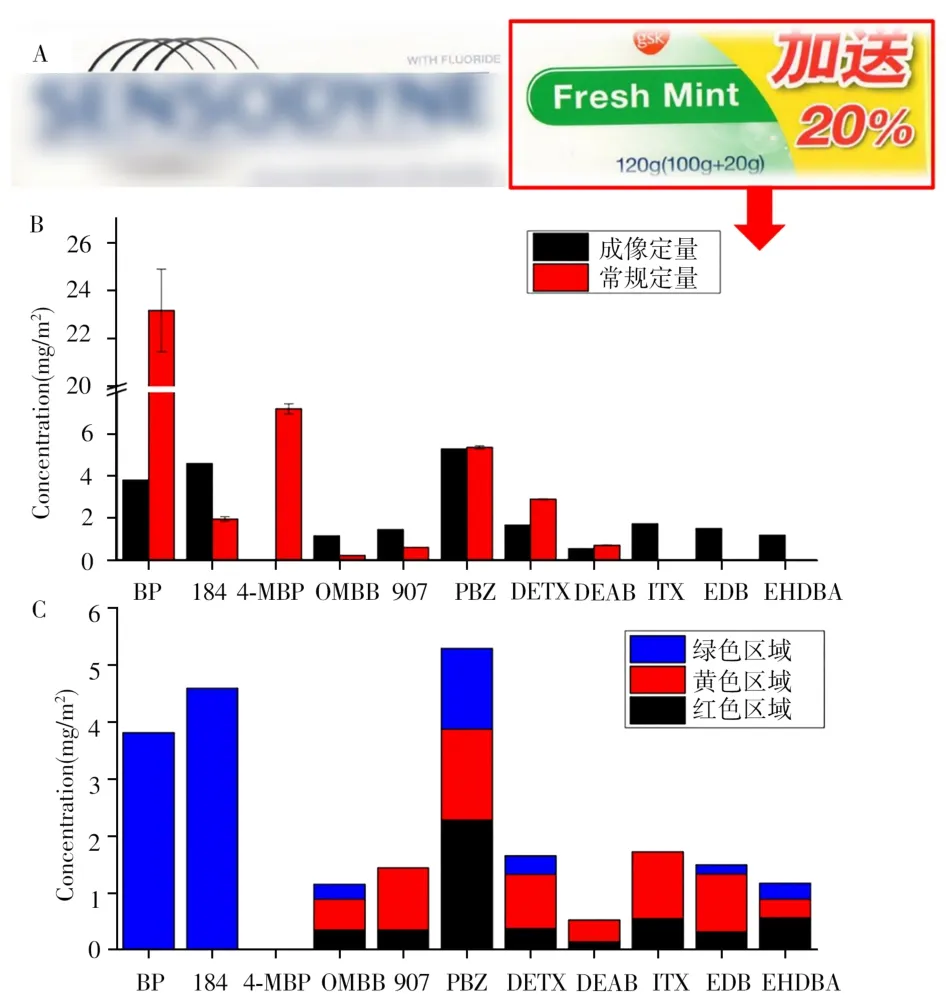
圖5 傳統GC-MS定量方法的取樣區域(A)、其與定量成像方法的結果比較(B)以及定量成像方法得到光引發劑在不同油墨區域的定量比例(C)Fig.5 The sampling area of the method of GC-MS(A),the comparison of the results with two methods(B)and the ratio of PIs in different ink areas obtained by the imaging method(C)
圖5C展示的是成像結果得到的紅框采樣區域3種不同顏色的油墨所對應的光引發劑含量占比。由圖可見,不同光引發劑在不同油墨區域中的占比有很大差別,而這個差別只能通過有空間分辨能力的成像技術才能顯示出來。
3 結 論
本研究利用微液節點采樣-質譜系統建立了包裝紙中光引發劑的定量成像方法,并將其用于包裝紙中多種光引發劑的檢測。結果表明,成像方法定量結果與傳統方法基本一致,且可以檢測到種類更多的光引發劑。另外,成像方法的空間分辨率還可提供傳統方法無法得到的光引發劑在包裝紙的空間分布信息,這對于禁用或限用光引發劑的溯源和檢測是非常有用的。另外,此方法對于指導和改進光固化油墨的涂布配方或工藝具有重大意義。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
