超高效液相色譜-串聯質譜法測定3種常見淋洗類化妝品中2種防腐劑
2021-11-25 07:28:52盧端萍程佳華謝宋陽
理化檢驗-化學分冊 2021年11期
盧端萍,陳 碩,程佳華,謝宋陽,王 勇
(福建省食品藥品質量檢驗研究院,福州 350001)
為了使化妝品在生產、使用和保存過程中免受微生物污染,保證化妝品的有效性,生產過程中往往需要加入防腐劑,甲基異噻唑啉酮(MI)和卡松由于水溶性較好,常用于洗發水、沐浴露、潔面乳等淋洗類化妝品中。卡松是甲基氯異噻唑啉酮(MCI)、MI與氯化鎂及硝酸鎂的混合物(MCI和MI質量比為3∶1)。MI和MCI具有較強的致敏性,長期使用含高添加量的MI和MCI的化妝品會引起接觸性皮炎[1-4]。歐盟(EU)2017/1224 公告對化妝品法規EC 1223/2009附錄V 進行修訂,規定MI僅可用于淋洗類化妝品,使用限量由0.01%降至0.001 5%。我國《化妝品安全技術規范》2015年版[5]中規定:化妝品中不能單獨使用MCI,MI最大允許添加量為0.01%;卡松僅能用于淋洗類化妝品,且不能和MI同時使用,最大允許添加量為0.001 5%。因此,有必要對化妝品中MI或卡松的含量進行監控。相關文獻中的MI和MCI測定方法主要有高效液相色譜法(HPLC)[6-11]、高效液相色譜-串聯質譜法(HPLC-MS/MS)[12-16]、氣相色譜法(GC)[17-18]、氣相色譜-質譜法(GC-MS)[19]。《化妝品安全技術規范》中規定的MI及MCI的測定方法為HPLC,兩種成分的測定下限均為0.001 3%(即13μg·g-1),與卡松的最大允許添加量0.001 5%(即15μg·g-1)接近,說明該方法的靈敏度較低,同時,MI和MCI的出峰時間較早,容易被樣品中極性較大的雜質干擾,無法被準確地測定。淋洗類化妝品中含有大量的表面活性劑,成分較為復雜,且卡松的添加量較低,因此需要建立一個高效、靈敏、準確的測定方法。本工作根據淋洗類化妝品的特性,對前處理、色譜條件、基質效應等進行考察,建立了超高效液相色譜-串聯質譜法(UHPLC-MS/MS)測定90批淋洗類化妝品中MI和MCI含量的方法,以期為化妝品中MI和MCI含量的準確測定提供參考。
1 試驗部分
1.1 儀器與試劑
ACQUITY UPLC I-Class型超高效液相色譜儀,配XEVO TQ-S型三重四極桿質譜儀;XP 205型電子天平。
單標準儲備溶液:稱取適量MI、MCI,加甲醇溶解并稀釋配制成40 mg·L-1的單標準儲備溶液。
混合標準溶液:分別移取適量MI、MCI單標準儲備溶液1,2 mL于100 mL容量瓶中,用10%(體積分數,下同)甲醇溶液稀釋配制成400,800μg·L-1的混合標準溶液。
混合標準溶液系列:移取適量混合標準溶液,用10%甲醇溶液逐級稀釋配制成含0.8,2,4,8,20,40,80,120,160,200μg·L-1MI和1.6,4,8,16,40,80,160,240,320,400μg·L-1MCI的混合標準溶液系列。
MI純度為98.6%;MCI純度為99.9%;甲醇為色譜純,其余試劑為分析純;試驗用水為一級水。
試驗樣品為近兩年福建省監督抽檢樣品,共90批,其中沐浴類產品48批(含嬰童類產品13批和沐浴洗發類產品1批)、洗發類產品33批(含嬰童類產品4批)、潔面類產品9批。
1.2 儀器工作條件
1.2.1 色譜條件
Waters ACQUITY UPLC HSS T3 色譜柱(100 mm×2.1 mm,1.8μm),柱 溫25 ℃;流量0.3 mL·min-1;進樣量1μL;流動相A 為甲醇,B為0.1%(體積分數,下同)甲酸溶液;按表1程序進行梯度洗脫。
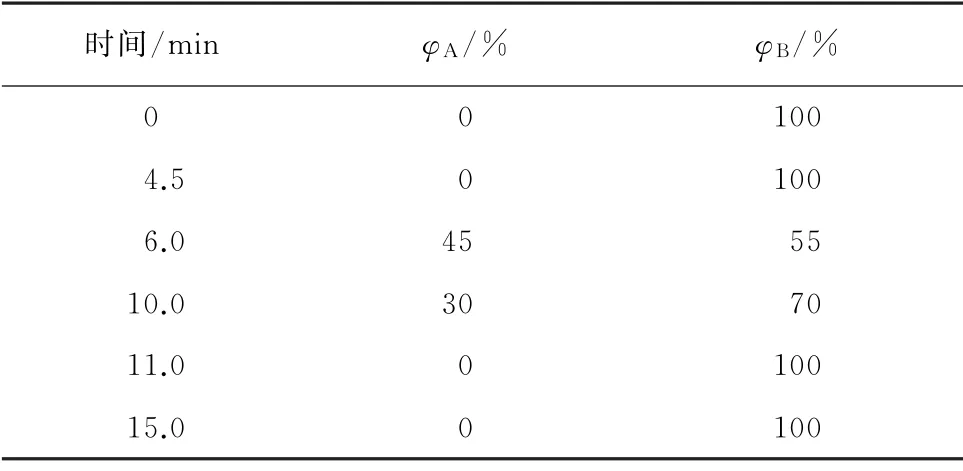
表1 梯度洗脫程序Tab.1 Gradient elution program
1.2.2 質譜條件
電噴霧離子源正離子模式(ESI+);毛細管電壓0.5 kV;脫溶劑溫度500 ℃;脫溶劑氣流量1 000 L·h-1,錐孔反吹氣流量150 L·h-1;霧化器壓力0.7 MPa。多反應監測(MRM)模式分析時間從第4.5 min開始,持續到第15 min;其他質譜參數見表2,其中,“*”為定量離子。
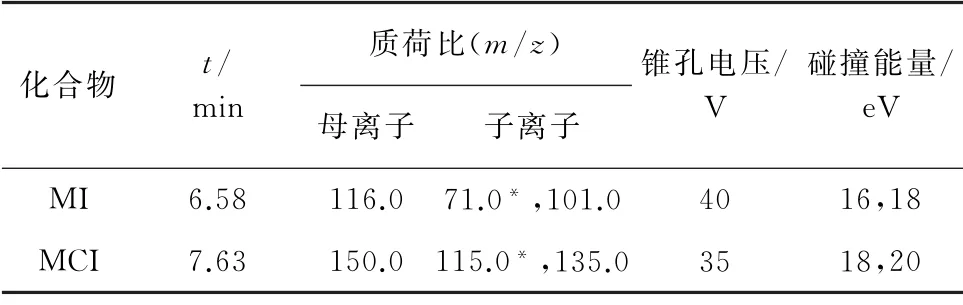
表2 質譜參數Tab.2 MS parameters
1.3 試驗方法
將0.2 g 樣品置于25 mL 納氏比色管中,加10%甲醇溶液1 mL,渦旋30 s。加入10%甲醇溶液7 mL,超聲提取10 min,依次加入100 g·L-1亞鐵氰化鉀溶液0.2 mL 和200 g·L-1乙酸鋅溶液0.2 mL,并分別混勻,用10% 甲醇溶液定容至10 mL,搖勻,靜置5 min,取上清液過0.22μm 濾膜,濾液按照儀器工作條件測定。
2 結果與討論
2.1 色譜行為
按照試驗方法分析含MI和MCI分別為40,80μg·L-1的混合標準溶液和3種不同類型的實際樣品,所得總離子流色譜圖見圖1。
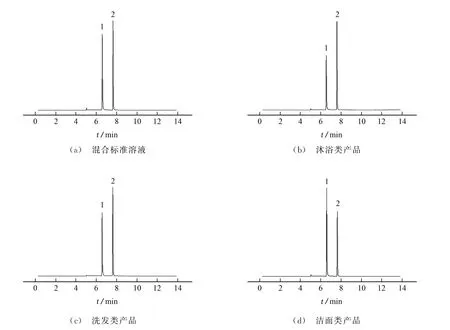
圖1 混合標準溶液和3種類型樣品的總離子流色譜圖Fig.1 Total ion chromatograms of mixed standard solution and 3 types of samples
2.2 色譜柱的選擇
試驗考察了 Waters BEH C18色譜柱(100 mm×2.1 mm,1.7μm)、Waters UPLC BEH HILIC色譜柱(100 mm×2.1 mm,1.7μm)、Waters CORTES T3色譜柱(100 mm×2.1 mm,2.7μm)、Waters ACQUITY UPLC HSS T3 色譜柱(100 mm×2.1 mm,1.8μm)、Agilent SB Aq色譜柱(100 mm×2.1 mm,1.8μm)、Agilent Poroshell HPH-C18色譜柱(150 mm×2.1 mm,2.7μm)對混合標準溶液中MI和MCI的分離效果。結果顯示:Waters UPLC BEH HILIC 色譜柱(100 mm×2.1 mm,1.7μm)所得MI和MCI出峰時間過早;Waters BEH C18色譜柱(100 mm×2.1 mm,1.7μm)、Waters CORTES T3 色 譜柱(100 mm×2.1 mm,2.7μm)所得MI出峰時間較早,MCI響應值較低;Agilent SB Aq色譜柱(100 mm×2.1 mm,1.8 μm)、Agilent Poroshell HPH-C18色譜柱(150 mm×2.1 mm,2.7μm)所得MI及MCI的色譜峰峰形均較差,且MCI 在Agilent Poroshell HPH-C18色譜柱(150 mm×2.1 mm,2.7μm)上出峰時間延遲較多;Waters ACQUITY UPLC HSS T3色譜柱(100 mm×2.1 mm,1.8μm)所得MI和MCI的出峰時間適當、色譜峰峰形較好、響應值較高,且該色譜柱耐水性好,故試驗選擇的色譜柱為Waters ACQUITY UPLC HSS T3 色譜柱(100 mm×2.1 mm,1.8μm)。
2.3 流動相的選擇
試驗分別以甲醇-0.1%甲酸溶液、甲醇-0.2%(體積分數,下同)甲酸溶液、含0.2%甲酸的甲醇溶液-0.1%甲酸溶液為流動相進行梯度洗脫,考察了不同流動相對混合標準溶液中MI和MCI分離效果的影響。結果顯示:用以上流動相進行梯度洗脫時,MI、MCI的峰形均較好,響應值均較高且相差不大。考慮到操作的簡便性,試驗選擇甲醇-0.1%甲酸溶液作為流動相。
2.4 樣品前處理條件的選擇
試驗曾嘗試先用飽和氯化鈉溶液分散樣品,再用乙腈萃取3 次,但在飽和氯化鈉溶液中檢出了MI,這是由于MI極性較大,無法完全轉移至乙腈層中,導致MI提取不完全。試驗又按照試驗方法提取樣品,并考察了不同體積分數(10%,30%,50%,100%)甲醇溶液作提取劑及不同超聲提取時間(5,10,15,20 min)對樣品中MI和MCI提取效果的影響。結果顯示:不同體積分數甲醇溶液的提取效果無明顯差別,且當超聲提取時間為10 min時,樣品中MI和MCI已被提取完全。從MI與MCI極性較大以及應減少有機溶劑使用的角度考慮,試驗選擇以10%甲醇溶液提取樣品,超聲提取時間為10 min。
2.5 沉淀劑用量的選擇
試驗以100 g·L-1亞鐵氰化鉀溶液和200 g·L-1乙酸鋅溶液作為沉淀劑沉淀樣品中的蛋白質等大分子雜質,并分別考察這2種沉淀劑用量分別為0.1,0.2 mL時的沉淀效果。結果顯示:沉淀劑的用量各為0.2 mL 時,離心后所得的沉淀較多,但MI、MCI的含量與0.1 mL時的一致。考慮到沉淀過程應盡可能除去較多雜質,試驗選擇2種沉淀劑的用量均為0.2 mL。
2.6 基質效應影響的消除
淋洗類化妝品中的基質比較復雜,表面活性劑等水溶性雜質容易產生基質效應,基質效應對質譜檢測影響較大。考慮到MI、MCI的極性較大,在反相色譜柱上不易保留,為了延長MI、MCI的保留時間,應適當提高水相的比例,試驗在梯度洗脫程序中,設置了先用0.1%甲酸溶液洗脫4.5 min這一步驟,以去除表面活性劑等水溶性雜質,且該時間段不進行質譜分析,從而降低基質效應。試驗還對比了HLB固相萃取柱凈化前后的基質效應,按照試驗方法分析實際樣品,并將得到的上清液過不同規格(60,100,200 mg)HLB固相萃取柱,結果顯示:MI在60,100 mg 的HLB 固相萃取柱上較容易被洗脫,在200 mg 的HLB 固相萃取柱上保留效果較好,但過柱所得的洗脫液中MI、MCI的含量與不過柱的上清液中的一致,說明是否過柱帶來的基質干擾對測定的影響較小,試驗選擇不凈化直接測定。
2.7 標準曲線、檢出限和測定下限
按照儀器工作條件測定混合標準溶液系列,以2種化合物的質量濃度為橫坐標,其對應的峰面積為縱坐標繪制標準曲線,線性參數見表3。
分別以3 倍、10 倍信噪比(S/N)計算檢出限(3S/N)和測定下限(10S/N),結果見表3。

表3 線性參數、檢出限和測定下限Tab.3 Linearity parameters,detection limits and lower limits of determination
2.8 重復性試驗
按照試驗方法平行測定一批沐浴類產品6次,計算測定值的相對標準偏差(RSD),MI和MCI的測定值分別為1.91,3.23μg·g-1,RSD 分別為0.70%,2.1%,表明該法重復性良好。
2.9 穩定性試驗
將按照試驗方法處理得到的樣品溶液在室溫下放置0,2,4,8,12,16,20,24 h后進樣,計算得MI、MCI峰面積的RSD 分別為2.1%,3.2%,說明樣品溶液在24 h內穩定。
2.10 回收試驗
按照試驗方法對空白沐浴露樣品進行3個濃度水平的加標回收試驗,每個濃度水平平行測定3次,計算回收率,結果見表4。
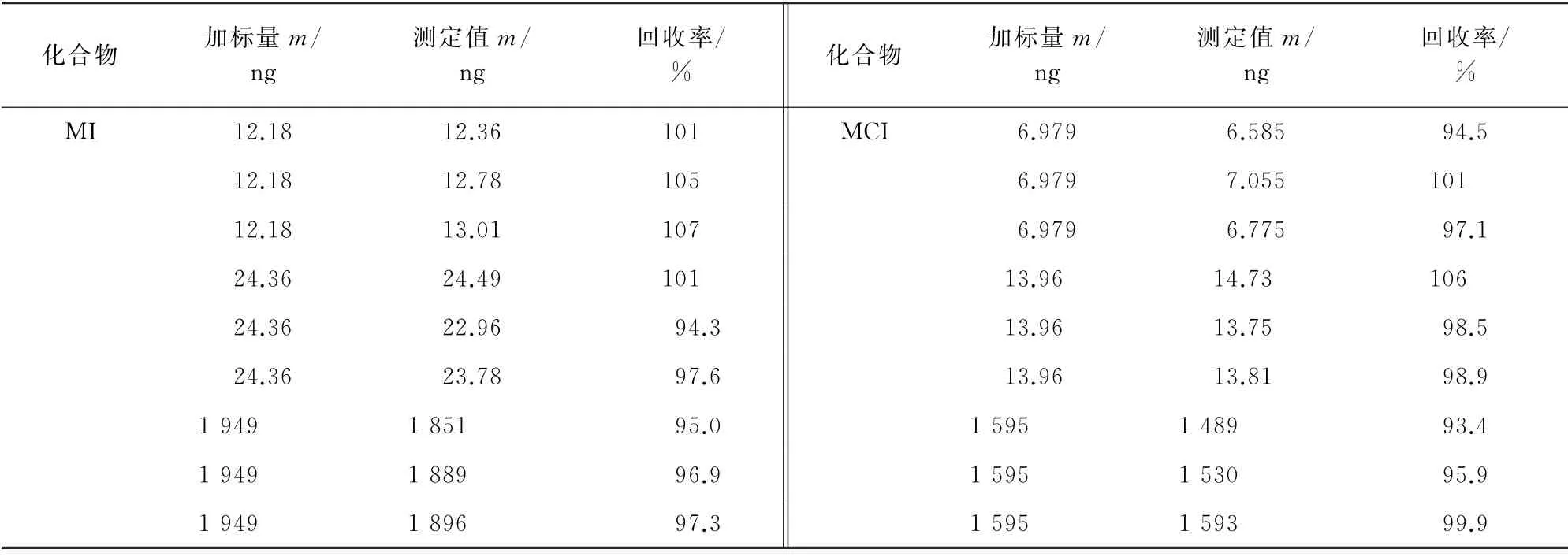
表4 回收試驗結果Tab.4 Results of test for recovery
由表4 可知:MI、MCI 的回收率分別為94.3%~107%和93.4%~106%,說明本方法的回收率較好。
2.11 樣品分析
按照試驗方法分析90批淋洗類化妝品,并與標簽標識符合情況進行比對,同時用《化妝品安全技術規范》中的HPLC 測定了抽檢的15 批產品中的MI、MCI含量,并同本方法進行比對,結果見表5。其中編號為61的樣品MI色譜峰附近有干擾峰,無法被準確測定。
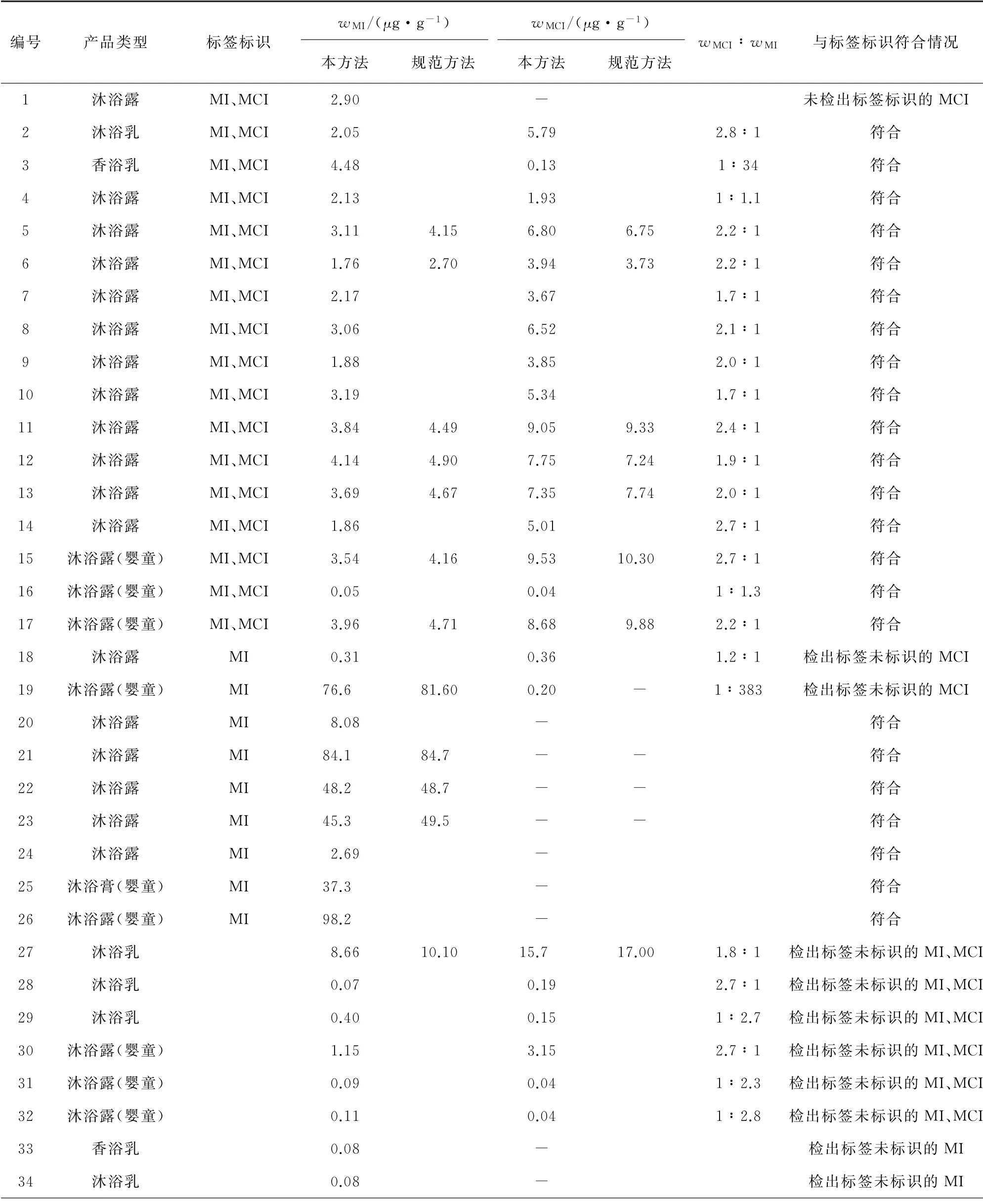
表5 樣品分析結果Tab.5 Analytical results of samples
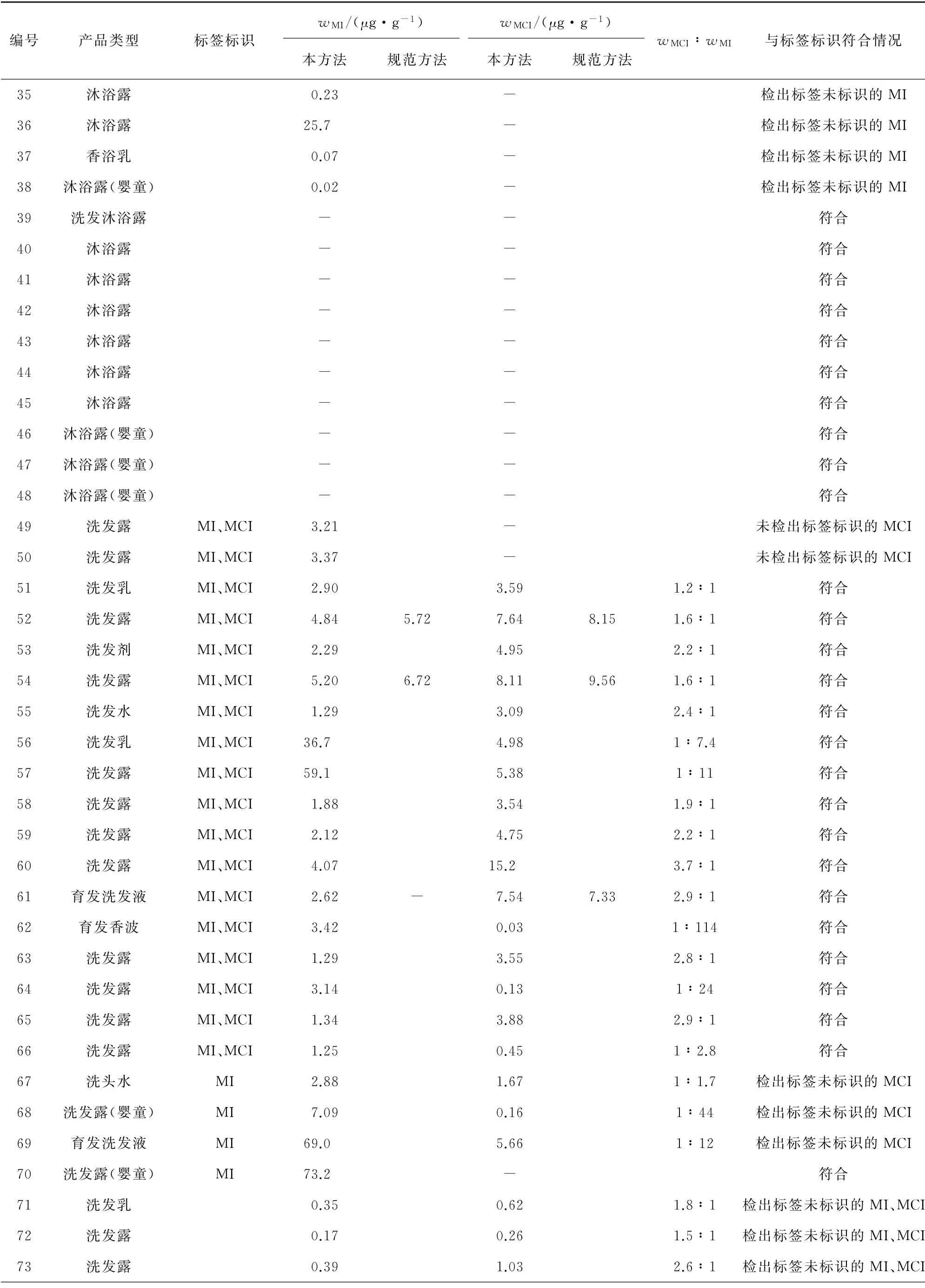
表5(續)

表5(續)
由表5可知,針對沐浴類產品:MI在成人、嬰童類產品中的檢出數量分別為28,10個,其含量超過歐盟化妝品法規EC 1223/2009 規定的限值(0.001 5%)的產品分別為4,3個,除1個嬰童類產品中MI的檢出量為98.2μg·g-1,接近我國《化妝品安全技術規范》規定的限值(0.01%)外,其他產品中MI含量均未超過限值;MCI在成人、嬰童產品中的檢出數量分別為17,7 個,最大檢出量分別為15.7,9.53μg·g-1。針對洗發類產品:MI在成人、嬰童類產品中的檢出數量分別為25,2個,其含量超過歐盟化妝品法規EC 1223/2009 規定的限值(0.001 5%)的產品分別為3,1 個,但均未超過《化妝品安全技術規范》規定的限值;MCI在成人、嬰童類產品中的檢出數量分別為21,1個,最大檢出量分別為15.2,0.16μg·g-1。針對潔面類產品:MI的檢出數量為3個,最大檢出量為2.67μg·g-1,未超過歐盟化妝品法規EC 1223/2009 及《化妝品安全技術規范》規定的限值;MCI的檢出數量為2個,最大檢出量為3.64μg·g-1。共有48批產品同時檢出MCI和MI,從二者的含量比的分布來看:33批產品MCI含量較MI的高,二者含量比為1.2∶1~3.7∶1,比值約為3∶1(2.6∶1~3.4∶1,依據GB/T 29666-2013《化妝品用防腐劑 甲基氯異噻唑啉酮和甲基異噻唑啉酮與氯化鎂及硝酸鎂的混合物》確定)的產品僅9批,符合《化妝品安全技術規范》卡松中MCI與MI質量比為3∶1的要求且未超過規定的限值(0.001 5%);15批產品中MI含量較MCI的高,MCI和MI的含量比為1∶1.1~1∶383,其中有8批產品MI含量比MCI高5倍以上,懷疑這些產品同時添加了卡松與MI。從與標簽標識符合情況來看,有25批(約28%)產品檢出情況與標簽標識的不一致:37批產品標簽同時標識了MI及MCI,但有3批產品未檢出MCI;14批產品標簽只標識了MI,但其中有5批產品還檢出了MCI;39批產品標簽未標識MI及MCI,但有9批產品同時檢出了MI及MCI,8批產品檢出了MI;涉及的產品包括沐浴類產品15批,洗發類產品10批。從方法比對結果來看:2種方法的測定值基本一致,但規范方法大多數測定值小于其測定下限(13.00μg·g-1)。
本工作建立了UHPLC-MS/MS測定淋洗類化妝品中MI和MCI的方法,并對實際樣品中MI及卡松的可能添加及與標簽標識符合情況進行分析。該方法準確度較高、專屬性較好、檢出限較低,可用于淋洗類化妝品中MI和MCI含量的測定。
