黑米中矢車菊色素的提取工藝優化及測定
2021-11-18 09:39:24廖若宇劉新保張春娥孫悅譚夢瑤
中國調味品 2021年11期
廖若宇,劉新保,張春娥*,孫悅,譚夢瑤
(1.寧夏回族自治區糧油產品質量檢測中心,銀川 750001;2.寧夏大學 化學化工學院,銀川 750001)
黑米,是黑稻加工而成的產品,外觀呈長橢圓形,素有“黑珍珠”和“世界米中之王”的美譽[1]。與普通白米相比,黑米具有良好的營養價值和功能屬性值,因其內部含有大量營養和生物活性成分,如必需氨基酸、功能性脂質、膳食纖維、維生素A和E、部分礦物質、花青素、酚類化合物、生育酚、植物甾醇和植酸等[2-5],故而引起越來越多的科研領域工作者的關注。
花青素,又稱花色素,是一種水溶性天然色素,屬酚類化合物中的類黃酮類物質[6],存在于植物細胞的液泡中。研究發現,現階段已知的花青素共有23種[7],其不僅能夠賦予植物鮮艷的色彩,而且能夠預防動脈粥樣硬化、降血糖、降血脂、抗疲勞、抗氧化及改善貧血等作用[8-10],在食品、藥品及保健品等多領域均得到廣泛應用。作為食品添加劑,可用于飲料、冰糕及果凍等產品的著色,而作為調味品,可用于調味料的著色和增香,如咖喱粉、芥末醬及火鍋底料等。因受自身結構及合成途徑的限制,花青素在光照、氧化劑、還原劑、防腐劑或鐵離子存在的環境中極易分解,致使穩定性大大降低。目前,已有學者對石榴皮[11]、桑葚[12-13]、紫薯[14]及黑豆[15]等植物中花青素的提取純化方式及功能活性成分進行研究,并且取得了不錯的研究成果。
本文選用黑米為原材料,以矢車菊素-3-O-葡萄糖苷為監測指標,農業部標準NY/T 3164-2017《黑米花色苷的測定 高效液相色譜法》[16]為依據,對標準方法中的提取方法及測定條件進行優化,改進后的方法在保證提取率的同時,能夠有效減少有毒有害試劑的用量及色素的提取成本,為今后黑米中花青素的提取提供了一種綠色高效的方法。
1 材料與方法
1.1 儀器與試劑
1.1.1 儀器
LC-20AT 液相色譜儀 島津國際貿易(上海)有限公司;XPE205 分析天平 梅特勒-托利多公司;SAH-B 水浴恒溫振蕩器 江蘇省金壇市環宇科學儀器廠;XC-300C 超聲波清洗機 濟寧鑫欣超聲電子設備有限公司;GL-20G-C高速冷凍離心機 上海安亭科學儀器廠;Lab Dancer 渦旋器 艾卡儀器設備有限公司;C300A 真空泵 德國Wgiiens公司;HMG-D24 氮吹儀 北京華安麥科生物技術有限公司;COMFORT純水/超純水機 德國賽多利斯公司。
1.1.2 材料與試劑
3種黑米產品:市購。
矢車菊素-3-葡萄糖苷標準品(Cy-3-glu):CAS號:7084-24-4,純度≥95%;95%乙醇(分析純):安徽安特食品股份有限公司;甲醇(色譜級):賽默飛世爾科技(中國)有限公司;鹽酸(優級純):永飛化學試劑有限公司;甲酸(色譜級):天津市光復精細化工研究所;Sep-Pak Vac 3cc NH2 Cartridges固相萃取柱(500 mg);5 mL一次性注射器:江蘇長城醫療器械有限公司;0.45 μm水相濾膜;0.45 μm有機相濾膜。
鹽酸溶液[c(HCl)=1 mol/L]:在200 mL容量瓶中加入適量去離子水,再緩慢加入濃鹽酸16.67 mL,邊加邊振搖,待冷卻至室溫后,定容至200 mL,搖勻。
提取液:95%乙醇和鹽酸溶液(1 mol/L)按照體積85+15混合,即取850 mL 95%乙醇和150 mL的鹽酸溶液混勻后備用。
流動相A:0.5%的甲酸水溶液,取5 mL甲酸,用去離子水定容至1000 mL,使用前過0.45 μm的水相濾膜,并脫氣。
流動相B:0.5%的甲酸甲醇溶液,取5 mL甲酸,用甲醇定容至1000 mL,使用前過0.45 μm的有機相濾膜,并脫氣。
試驗用水均為去離子水。
1.2 試驗方法設計
1.2.1 標準品溶液的配制
1.2.1.1 標準儲備液的配制
精確稱量2.5 mg矢車菊素-3-葡萄糖苷標準品于25 mL棕色容量瓶中,提取液定容至刻度,即可得到濃度為100 μg/mL的標準儲備液。
1.2.1.2 標準曲線點的配制
取1 mL矢車菊素-3-葡萄糖苷標準儲備液于10 mL棕色容量瓶中,提取液定容至刻度,得到10 μg/mL的標準中間儲備液。再分別從中間儲備液中吸取適量溶液于1 mL的進樣瓶中,再次用提取液定容,得到濃度分別為1,2,5,10,20,50,100 μg/mL的系列標準工作溶液。
1.2.2 色譜條件確定
色譜柱:Inertsil ODS-SP C18(250 mm×4.6 mm,5 μm),日本島津公司;檢測波長520 nm,柱溫:30 ℃,流動相:0.5%的甲酸水溶液和0.5%的甲酸甲醇溶液(45∶55,V/V),流速:1.0 mL/min;進樣體積10 μL,等濃度洗脫,分析時間設定為10 min,約6 min左右開始出峰。
1.2.3 樣品前處理
準確稱量0.50 g(精確至0.01 g)粉碎細度達80~100目且混合均勻的樣品,置于50 mL離心管中,向其中加入15.00 mL矢車菊色素提取液,混勻后在80 W功率下超聲5 min,隨后在70 ℃恒溫振蕩器上中速振蕩25 min,靜置冷卻至室溫,然后以4000 r/min離心5 min,轉移上清液至50 mL比色管中。再向離心管中加入15.00 mL提取液進行二次提取,渦旋20 s,重復上述操作,合并兩次提取液,避光保存待測。
1.2.4 樣品凈化
先用6 mL甲醇活化凈化柱,棄去濾液;再取1 mL待測液上樣,棄去樣液;隨后以5 mL洗脫液(甲醇∶甲酸為1∶1,V/V)洗脫固相萃取柱,收集洗脫液于10 mL小試管中,在45 ℃下氮吹儀上濃縮至干,最后用1 mL提取液復溶,渦旋混合1 min,待樣品充分溶解后,過0.22 μm尼龍針式過濾器,待上機分析。
1.2.5 單因素考察及綜合因素優化
選取單因素比對法,對試驗過程中的黑米粉粗細度、提取方式、提取次數、提取液種類、料液比、水浴振蕩提取溫度、水浴提取時間、凈化柱8個因素分別考察,依據單因素優化出的結果選取料液比、振蕩提取時間及水浴振蕩提取溫度3個因素,采用Box-Behnken設計后續優化試驗,響應面因素水平見表1。以測得的黑米樣品中的含量為響應值。同時通過Design-Expert 8.0.6分析軟件得到二次回歸方程和誤差分析,確定各因素數值的最優組合并對其進行驗證。

表1 響應面試驗因素水平表Table 1 The factors and levels of response surface experiment
2 結果分析
2.1 單因素試驗結果
2.1.1 黑米粉粗細度對矢車菊素含量的影響
分別對比20,40,60,80,100目及整米6種不同樣品對矢車菊色素含量的影響(見圖1),樣品前處理方法參照1.2.3,測定方法見1.2.2。
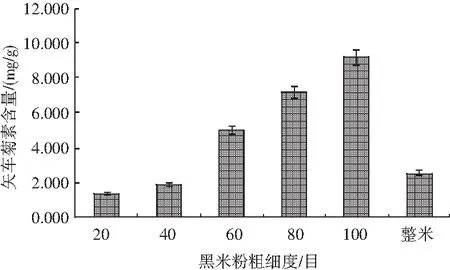
圖1 黑米粉粗細度對矢車菊素含量的影響Fig.1 Effect of the particle size of black rice flour on the content of cyanidin
試驗發現,樣品粉碎后過不同目數的篩網時,隨著黑米粉顆粒度越來越細,樣品中的色素含量越來越高,并且混勻后的樣品顏色與樣品顆粒度呈現負相關,即顆粒度越細,混勻后的樣品顏色越深。同時在過80目和100目篩網時,有許多肉眼可見的黑色“斑點”存在,從而影響了整個黑米粉的顏色。同樣是粉碎細度為100目,直接粉碎后的樣品并沒有過篩后的顏色深,由此推斷在篩不同目數的黑米樣品時,小顆粒度的黑米種皮可能落到靠近底層的篩網中,因此黑米中的色素可能主要存在于種皮中。對黑米種皮、碾后黑米兩種不同樣品中的矢車菊色素含量進行測定,結果見圖2。
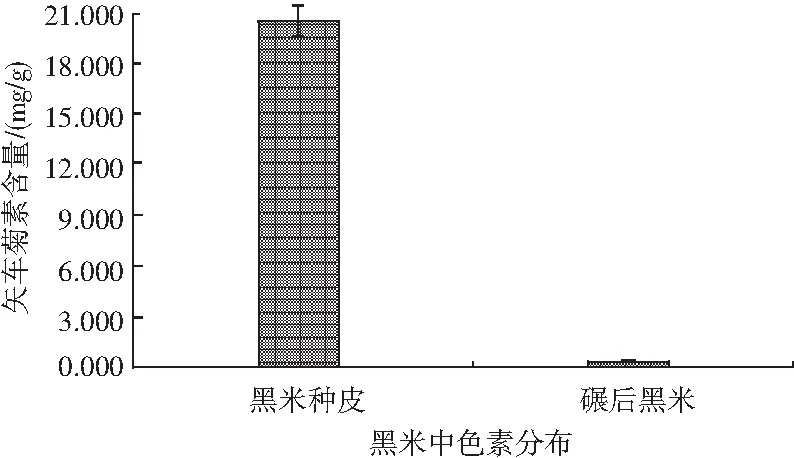
圖2 黑米中色素的分布Fig.2 The distribution of pigment in black rice
由圖2可知,黑米中的矢車菊色素確實主要存在于黑米種皮中,而碾后黑米僅含有少量色素。同時通過分析試驗數據可得黑米種皮中色素含量是碾后黑米的130倍以上。本著以節約資源為目的,同時考慮到實際操作的便利性,本設計選取直接過錘式旋風磨后細度達100目的樣品進行后續試驗。
2.1.2 提取方式對矢車菊素含量的影響
分別對比振蕩30 min、渦旋15 min、超聲30 min及超聲加振蕩共計30 min(超聲5 min,振蕩25 min)4種提取方式對色素含量的影響,見圖3。
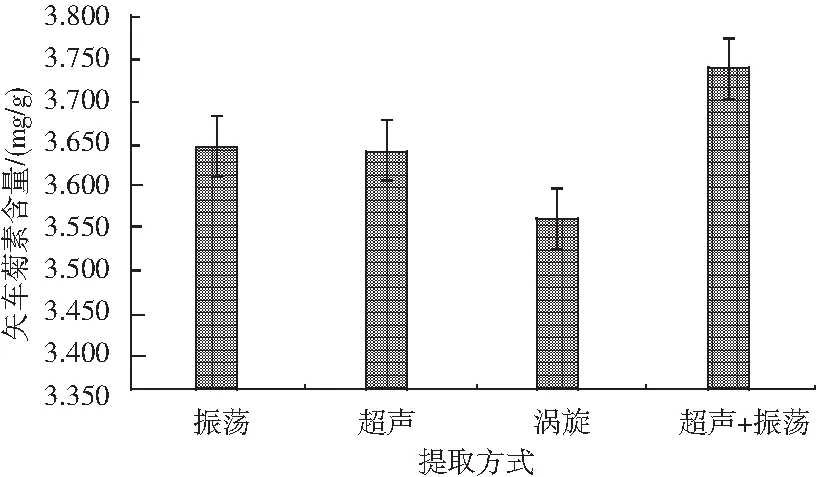
圖3 提取方式對矢車菊素含量的影響Fig.3 Effect of extraction methods on the content of cyanidin
樣品前處理方法參照1.2.3,測定方法見1.2.2。試驗發現,振蕩及超聲兩種提取方式對色素的提取影響基本相近,渦旋提取效果最差,超聲加振蕩兩種方式混合提取后,樣品中矢車菊色素含量較高,因此后續試驗中均選取超聲和振蕩兩種方式混合提取。
2.1.3 提取次數對矢車菊素含量的影響
對比不同提取次數對矢車菊色素含量的影響,見圖4。樣品前處理方法參照1.2.3,測定方法見1.2.2。
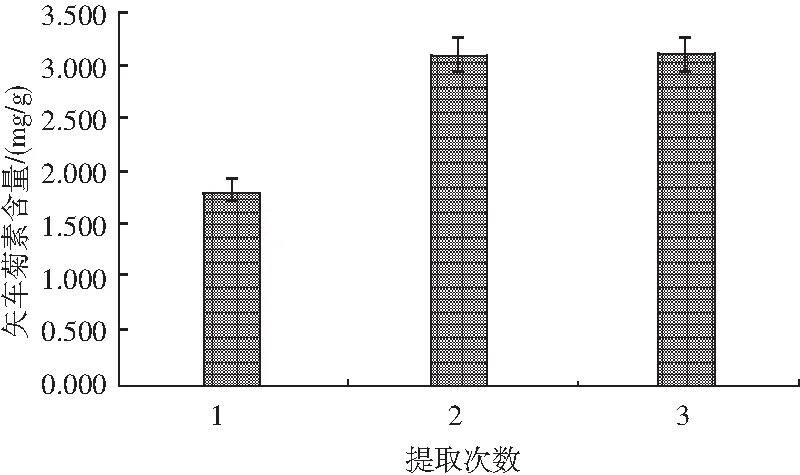
圖4 提取次數對矢車菊素含量的影響Fig.4 Effect of extraction times on the content cyanidin
試驗發現,當提取次數從1次增加至2次時,色素含量顯著提升,但提取3次時色素含量與提取2次差別不大。由此說明在一定范圍內增加提取次數能夠使樣品中色素被充分提取出來,但并不是提取次數越多越好。因此,本試驗選取提取2次最佳。
2.1.4 不同提取試劑對矢車菊素含量的影響
通過查閱文獻和資料,得知矢車菊色素易溶于甲醇、乙醇及丙酮,可溶于水。由此推斷該色素易溶于有機試劑,遂隨機選取95%乙醇、二氯甲烷、石油醚、丙酮、正己烷、鹽酸與甲醇混合液、鹽酸與95%乙醇混合溶液、鹽酸與水混合溶液共計8種不同提取試劑提取黑米中的矢車菊色素,見圖5。
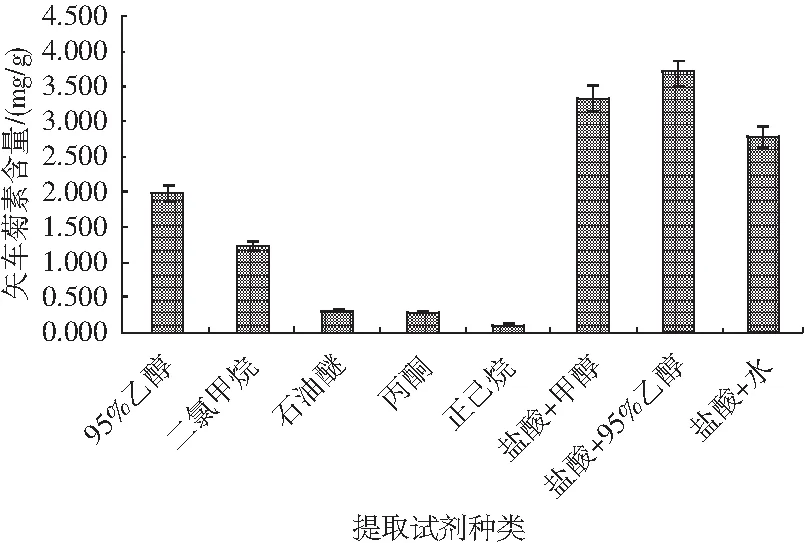
圖5 不同提取試劑對矢車菊素含量的影響Fig.5 Effect of different extraction reagents on the content of cyanidin
稱取16份0.50 g樣品,置于50 mL離心管中,分別向其中加入15.00 mL不同提取液,每種提取液設置兩個平行,混勻后在80 W功率下超聲5 min,隨后在65 ℃恒溫振蕩器上中速振蕩25 min,靜置冷卻至室溫,然后以4000 r/min離心5 min,轉移上清液至50 mL比色管中。重復上述操作2次,待上機測試分析。測定結果表明,依據農業標準NY/T 3164-2017《黑米花色苷的測定 高效液相色譜法》中鹽酸與甲醇混合液以及鹽酸與95%乙醇混合溶液提取效果較好,但考慮提取效果最佳、試劑毒性較小,首選鹽酸與95%乙醇混合溶液作為后續試驗的提取溶液。
2.1.5 不同料液比對矢車菊素含量的影響
選取鹽酸與95%乙醇混合溶液為提取液,以農業標準中的鹽酸與甲醇混合液作參考,分別對比不同料液比下黑米中矢車菊色素含量,見圖6。
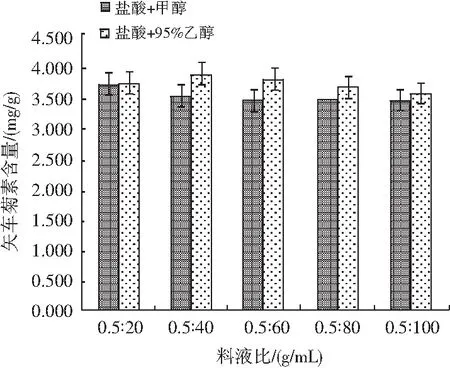
圖6 不同料液比對矢車菊素含量的影響Fig.6 Effect of different solid-liquid ratios on the content of cyanidin
稱取20份0.50 g樣品,置于50 mL離心管中,分別向其中加入10,20,30,40,50 mL的提取液,每個水平設置兩個平行,混勻后在80 W功率下超聲5 min,隨后在65 ℃恒溫振蕩器上中速振蕩25 min,靜置冷卻至室溫,然后以4000 r/min離心5 min,轉移上清液至體積適量的比色管中。重復上述操作2次,待上機測試分析。結果顯示,料液比為0.5∶40時,兩種提取液提取色素的效果均優于其他,同時鹽酸與95%乙醇混合溶液作為提取液時,效果明顯優于標準中提取液的提取效果。因此,本試驗選取鹽酸與95%乙醇混合溶液作為提取試劑,提取試劑添加量為40 mL。
2.1.6 不同振蕩溫度對矢車菊素含量的影響
考察不同溫度對矢車菊色素含量的影響,見圖7。
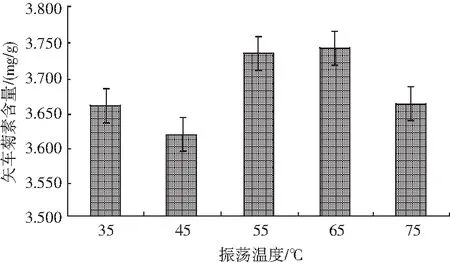
圖7 不同振蕩溫度對矢車菊素含量的影響Fig.7 Effect of different oscillation temperatures on the content of cyanidin
稱取10份0.50 g樣品,置于50 mL離心管中,加入20.00 mL提取液,混勻后在80 W功率下超聲5 min,隨后分別在35,45,55,65,75 ℃恒溫振蕩器上中速振蕩25 min,每個水平兩個平行,靜置冷卻至室溫,然后以4000 r/min離心5 min,轉移上清液至50 mL比色管中。重復上述操作2次,待上機測試分析。結果顯示:在溫度為65 ℃下,恒溫振蕩提取黑米中的矢車菊色素效果較好。因此,本試驗選擇提取溫度為65 ℃進行后續試驗。
2.1.7 不同提取(超聲提取+恒溫振蕩提取)總時間對矢車菊素含量的影響
考察不同提取總時間對黑米中矢車菊色素含量的影響,見圖8。
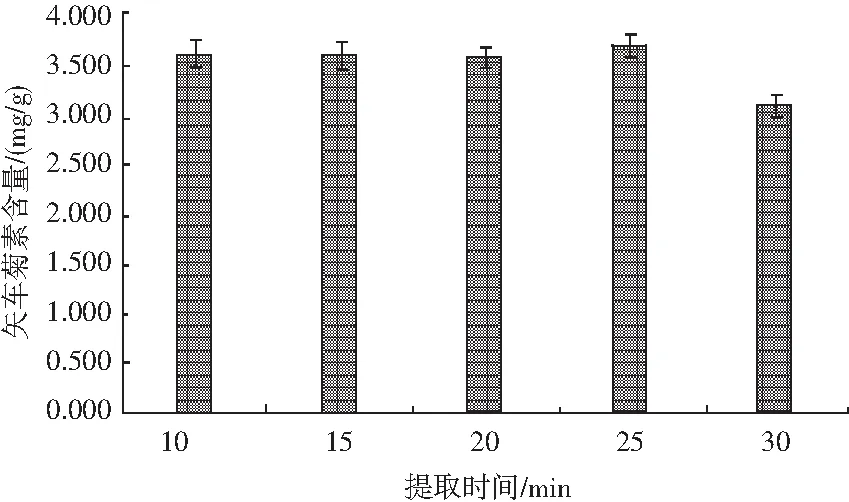
圖8 不同提取總時間對矢車菊素含量的影響Fig.8 Effect of different extraction total time on the content of cyanidin
稱取10份0.50 g樣品,置于50 mL離心管中,加入20.00 mL提取液,混勻后在80 W功率下超聲5 min,隨后分別在65 ℃恒溫振蕩器上中速振蕩5,10,15,20,25 min,靜置冷卻至室溫,然后以4000 r/min離心5 min,轉移上清液至50 mL比色管中。重復上述操作2次,待上機測試分析。結果顯示:當超聲提取時間固定為5 min,振蕩提取時間為5~15 min內,黑米中提取的矢車菊色素含量差別不大,當提取時間增加至20 min時,提取效果略優于其他幾個條件。因此,本試驗選取總提取時間為25 min,即超聲5 min,振蕩20 min。
2.1.8 不同凈化柱的選擇對矢車菊素含量的影響
由于黑米樣品中存在矢車菊素-3-O-葡萄糖苷和芍藥素-3-O-葡萄糖苷2種花色苷,本試驗測定矢車菊色素時樣品基線不穩,猜測可能是芍藥色素對其產生了干擾,為避免長時間測定對后續樣品產生干擾及污染色譜柱,嘗試選擇利用固相萃取柱凈化樣品。稱取12份0.50 g樣品,置于50 mL離心管中,向其中加入20.00 mL矢車菊色素提取液,混勻后在80 W功率下超聲5 min,隨后在65 ℃恒溫振蕩器上中速振蕩20 min,靜置冷卻至室溫,然后以4000 r/min離心5 min,轉移上清液至50 mL比色管中。重復上述操作2次,按照1.2.4的方法凈化樣品,分別選取PWAX、Strata、C18、FL、NH2、3cc 6種凈化柱凈化同一樣品,每種凈化柱設置兩個平行,試驗結果見圖9。
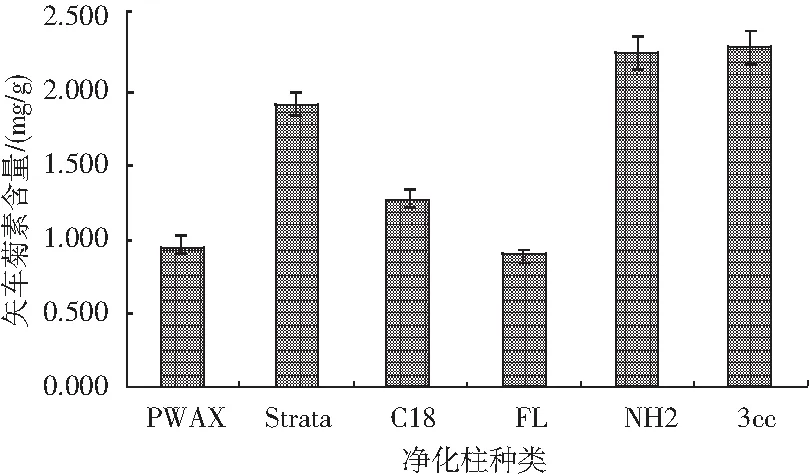
圖9 不同凈化柱的選擇對矢車菊素含量的影響Fig.9 Effect of different purification columns on the content of cyanidin
結果表明,NH2和3cc兩種小柱的凈化效果明顯好于其他種類的凈化柱,并且所選擇的6種凈化柱中,3cc最優。因此,后續試驗中選擇最佳凈化柱3cc。
選取3cc凈化柱,活化小柱后,分別添加2%甲酸水及25%氨水甲醇,制造酸性及堿性環境條件,并在此條件下對樣品進行凈化以測定色素含量,試驗結果見圖10。
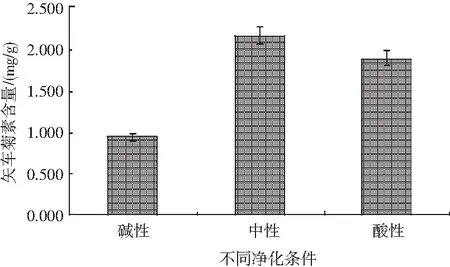
圖10 不同酸堿條件下凈化柱對黑米中矢車菊素凈化效果比對Fig.10 Comparison of purification columns on purification effects of cyanidin in black rice under different acid and alkali conditions
試驗發現,在酸性環境中,樣品隨著上樣后樣液流出而流失,無法留存于凈化柱上,造成樣品中色素含量降低。堿性環境與酸性環境類似,但流失量較酸性環境少一些,推測可能是由于色素提取過程中的部分酸與活化凈化柱后添加的堿液反應,從而造成色素流失。因此,本試驗選取中性條件,即活化固相萃取小柱后直接上樣的方式凈化樣品。
2.2 響應面試驗
2.2.1 建立二次回歸方程及誤差分析
對表2中的數據進行回歸擬合得到二次回歸方程為:矢車菊素含量=3.60238+0.033800A-0.11970B+9.50000E-3C+4.45000E-4AB-6.85000E-4AC+1.59000E-3BC。
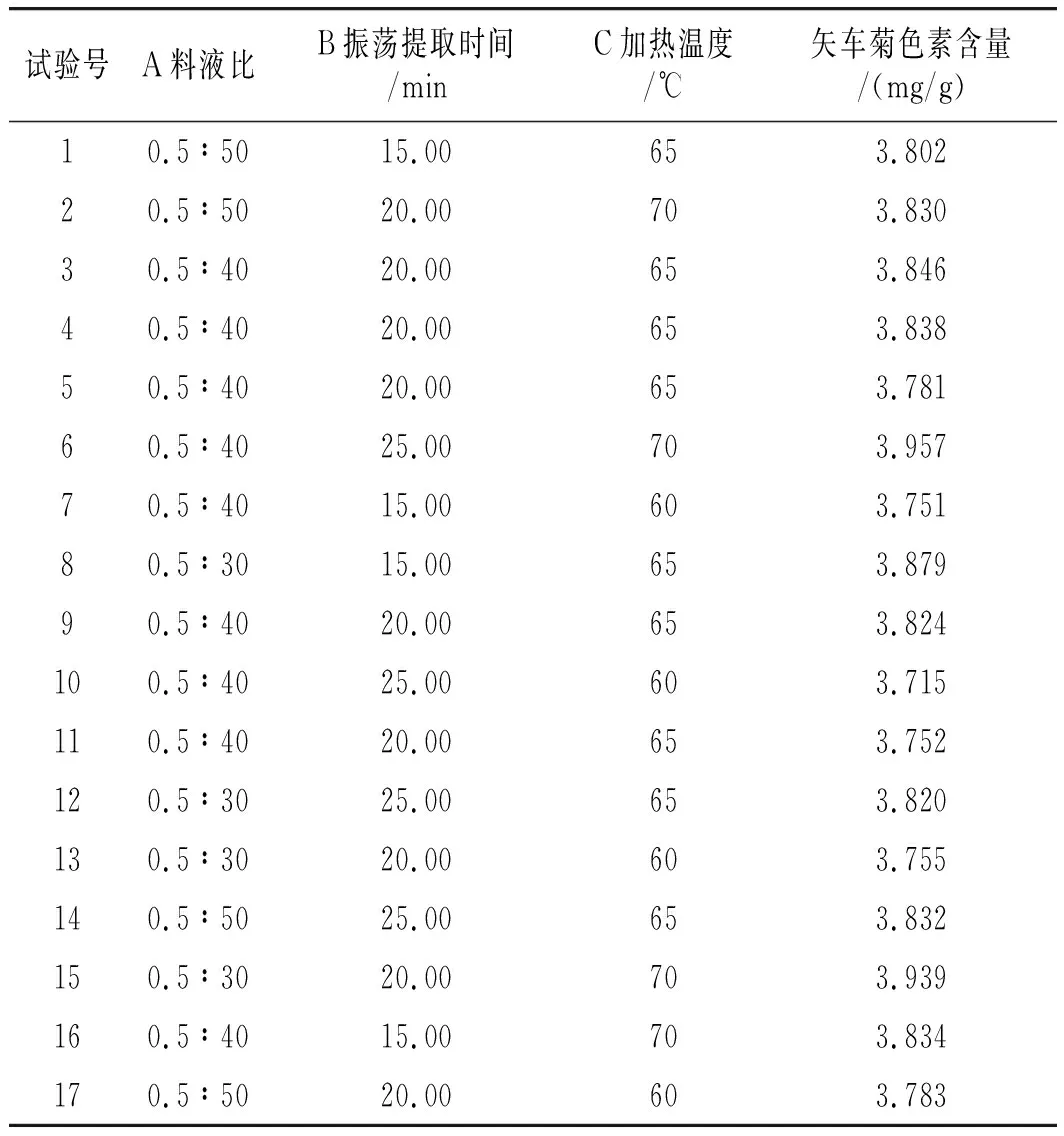
表2 響應面試驗設計與結果Table 2 The design and results of response surface test
通過ANOVA進行回歸方程誤差分析,結果見表3。
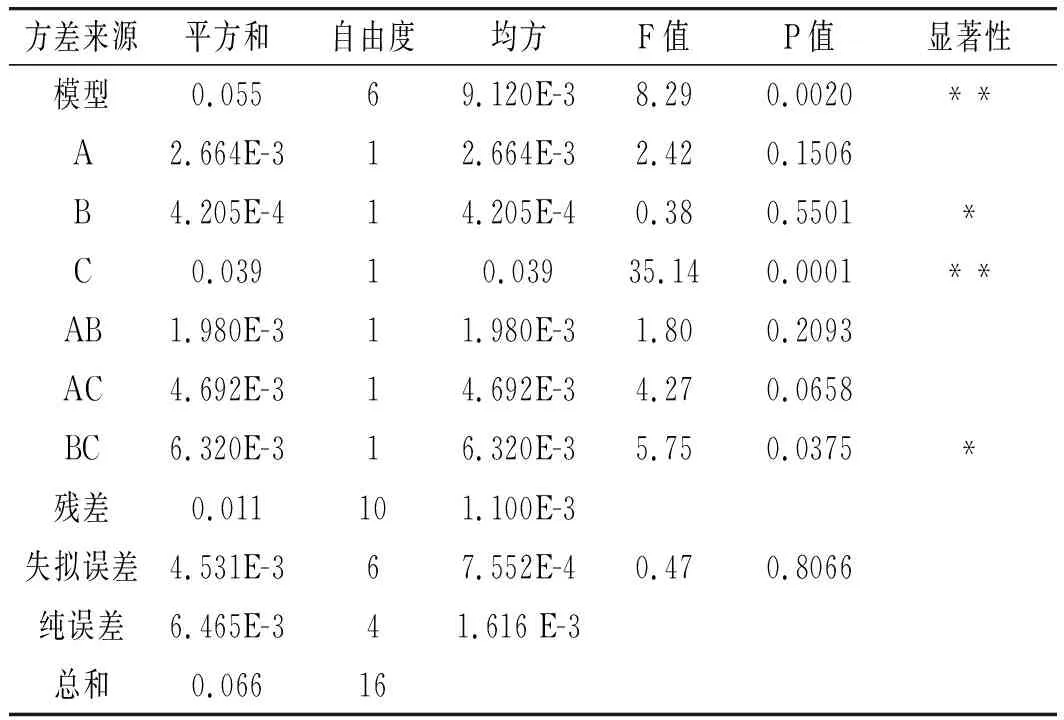
表3 響應面試驗方差分析結果Table 3 The variance analysis results of response surface test
由表3可知,該模型回歸顯著,失擬項不顯著,模型CV=0.87%,R2=0.8327,RAdj2=0.7323,其中CV值與模型置信度呈現負相關,而相關系數R2和RAdj2越接近1,模型相關性則越好。本試驗中數據表明該模型與實際試驗擬合較好,自變量與響應值之間線性關系顯著,可以用于黑米中矢車菊色素的提取與含量預測。另外,所考察的3個因素中,C對黑米中矢車菊色素提取的影響高度顯著;B、BC的影響顯著;A、AB、AC的影響不顯著,各因素影響從大到小依次為振蕩提取時間>料液比>加熱溫度。
2.2.2 優化組合驗證試驗
為確定最佳提取方式,使用快速上升法進行優化,得到提取黑米中矢車菊色素的最佳方案為:料液比0.5∶30,振蕩提取時間25.00 min,振蕩提取加熱溫度70.00 ℃,預估黑米中矢車菊色素含量為3.967 mg/g。對上述條件進行試驗結果的驗證,結果見表4。

表4 最優組驗證試驗Table 4 The validation test of optimal group
由表4可知,最優組矢車菊色素提取含量為3.972 mg/g,達到了理論預測值,同時兩者相對誤差約為0.005 mg/g,表明響應面軟件優化后得出的回歸方程有一定的實踐指導意義。
2.3 標準曲線的測定
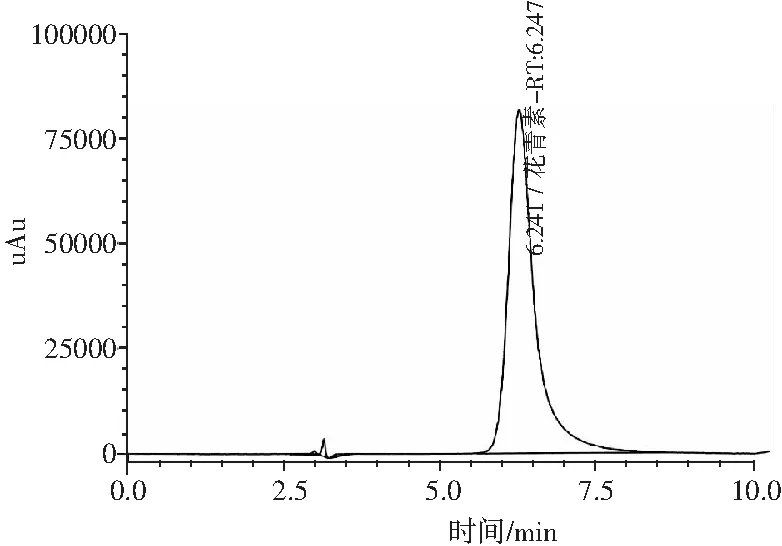
圖11 矢車菊色素標準品譜圖Fig.11 The spectrum of cyanidin pigment standard sample
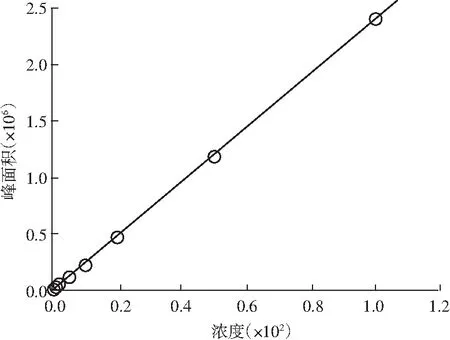
圖12 矢車菊色素標準曲線圖Fig.12 The standard curve of cyanidin pigment
以1.2.1.2的方法配置各個標準曲線點,以1.2.2 色譜條件上機測定。矢車菊色素標品譜圖見圖11。以濃度為橫坐標,對應的色譜峰面積為縱坐標,繪制標準曲線,建立線性回歸方程,見圖12。當矢車菊色素在1~100 μg/mL范圍內時,濃度與峰面積呈現良好的線性關系,回歸方程為f(x)=24040.5x-10797.8,R=0.9999。最低檢出限為0.5 μg/mL,定量限為2 μg/mL。
2.4 樣品含量測定
隨機購買市面上3種不同品牌的黑米樣品,對其矢車菊色素含量進行測定,測定結果見表5。

表5 樣品中矢車菊色素含量Table 5 The content of cyanidin pigment in samples
由表5可知,3種不同黑米樣品,未凈化時內部矢車菊色素含量在2.953~3.487 mg/g之間,相對標準偏差為0.470%~3.590%,而經過凈化的樣品,其矢車菊色素含量在2.596~3.452 mg/g之間,相對標準偏差為0.134%~4.229%,證明經過凈化后的樣品,矢車菊素含量損失在0.5 mg/g范圍內,可根據實際需求,選擇是否需要凈化樣品。
3 結論
本文在傳統液相色譜法測定的基礎上,結合使用固相萃取小柱凈化樣品。經驗證,相比于農業部標準NY/T 3164-2017《黑米花色苷的測定 高效液相色譜法》,本方法更加綠色環保,在保證提取率的同時,有效減少了有機試劑的污染及提取成本,為黑米中矢車菊色素的提取提供了一種可靠方法。本試驗研究得出當料液比為0.5∶15,超聲提取時間為5.00 min,振蕩提取時間為25.00 min,振蕩提取加熱溫度為70.00 ℃,重復2次提取時,能夠有效提取黑米中的矢車菊色素,測得其含量為3.972 mg/g,相對標準偏差為1.54%。同時,利用Sep-Pak Vac 3cc NH2 Cartridges固相萃取柱凈化后的樣品雜質干擾小、分離度好,能夠更加準確地定量出黑米樣品中的矢車菊色素含量。綜上所述,本文為黑米中天然色素的提取、凈化及測定提供了一種新的、快速、可靠的方法。
