超高效液相色譜-串聯質譜法測定畜禽毛發中4種違禁氟喹諾酮類獸藥
2021-11-15 05:37:04王夢芝周瑤敏胡麗芳鄭小明周日龍閔佳玲余云云
分析科學學報 2021年5期
關鍵詞:檢測
費 丹, 王夢芝, 周瑤敏, 謝 敏, 胡麗芳, 鄭小明,周日龍, 閔佳玲, 余云云, 徐 俊*
(1.江西省農業科學院農產品質量安全與標準研究所,江西省農產品質量安全重點實驗室,江西南昌 330200;2.揚州大學動物科學與技術學院,江蘇揚州 225009;3.江西省玉山黑豬原種場,江西上饒 334700)
氟喹諾酮類藥物(Fluoroquinolones,FQS)是在喹啉環的6位上加入了氟原子,7位上連有哌嗪基的一類衍生物[1,2]。在獸醫臨床上,常作為細菌感染類疾病的治療藥物,具有抗菌譜廣、高效、低毒、促生長等優點[3,4]。然而,在動物養殖過程中經常出現超劑量、超范圍使用,使得細菌產生耐藥性[5]。與此同時,氟喹諾酮類藥物殘留造成的食品安全問題層出不窮,殘留有藥物的畜禽產品通過食物鏈傳遞可能對人體產生一系列危害,如造成中樞神經系統紊亂,使人出現失眠、頭痛及驚厥等癥狀,嚴重的還會引發光毒性、肝腎毒性和溶血性貧血等疾病[6]。鑒于此,中國農業農村部于2015年發布了2292號公告,要求停止在食品動物中使用洛美沙星、培氟沙星、氧氟沙星和諾氟沙星這4種獸藥。然而,近幾年風險評估研究發現這4種違禁藥物依然在養殖過程中違規使用,尤其是養殖戶為了逃避農產品質量安全監管,常在養殖前端過程中使用,而畜禽在屠宰上市時由于藥物自身代謝作用,常規的快檢設備無法檢出藥物殘留,這無形中給畜禽產品養殖全過程的質量安全監管帶來了難題。
當前,氟喹諾酮類藥物的分析檢測手段主要包括分光光度法、放射免疫法(RIA)、微生物法、酶聯免疫吸附法(ELISA)、電解分析法、高效液相色譜-質譜法(HPLC-MS/MS)等[7 - 11]。其中,液相色譜-串聯質譜法尤其對復雜樣品具有較高的分離能力,并且選擇性強、靈敏度高,能夠提供相對分子量和結構信息,因此是目前獸藥殘留等痕量分析的主要方法[12,13]。由于氟喹諾酮類抗生素特殊的化學結構,容易生成正離子,因此流動相中常添加甲酸,以此來提高離子化效率。檢測基質主要包括畜禽肌肉組織、雞蛋、血漿、內臟或獸藥等[14 - 16]。其中,肌肉、內臟、血液需要通過屠宰動物的方式來獲取樣品,其成本高、步驟繁瑣。收集畜禽毛發樣品不僅可以避免動物被屠宰,并且取樣簡便、易于保存;此外,毛發組織中沒有血液循環和降解藥物的活性物質以及高效的排泄方式,因此藥物的代謝緩慢,并且在毛發中的殘留時間比其他組織更長,有利于養殖過程中對違禁藥物使用的高效監管[17]。
目前,已有關于人頭發中農藥[18]殘留檢測,以及畜禽毛發中利巴韋林[19]、氟蟲腈[20]等藥物殘留檢測的研究報道,而有關畜禽毛發中培氟沙星、氧氟沙星、諾氟沙星和諾美沙星4種違禁氟喹諾酮類藥物的檢測方法鮮有報道。本研究通過乙腈-乙酸混合提取,采用PSA/C18凈化管進行基質分散凈化,Agilent ZORBAX Eclipse Plus C18色譜柱分離,電噴霧串聯質譜多反應監測(MRM)模式測定,建立了畜禽毛發中上述4種違禁氟喹諾酮類獸藥殘留量的檢測方法,為畜禽產品在養殖過程中的高效監管提供技術支撐。
1 實驗部分
1.1 儀器與試劑
1290-6495超高效液相色譜-串聯質譜儀(美國Agilent公司);MS205DU電子分析天平(瑞士,Mettler Toledo公司);GL-88B型旋渦混合器(海門市Qilinbeier公司);KH-500DB型數控超聲波清洗器(昆山HeChuang Ultrasonic公司);電熱恒溫干燥箱(上海HeDe Laboratory公司);離心機(上海SH-AnTing公司);N-EVAP112氮吹儀(美國,Organomation公司);0.22 μm尼龍濾膜(美國,Agilent公司);Milli-Q超純水器(Millipore公司);固相萃取裝置(美國,Supelco公司)。
培氟沙星、氧氟沙星、諾氟沙星、洛美沙星、培氟沙星-D5、氧氟沙星-D3、諾氟沙星-D5、洛美沙星-D5(美國Sigma-Aldrich公司);十八烷基硅烷(C18,粒徑40~60 μm,上海阿拉丁公司);色譜純甲酸、甲醇、乙腈(美國Fisher試劑公司);十二烷基硫酸鈉(SDS,上海Macklin公司);乙二胺-N-丙基硅烷(PSA,粒徑40~60 μm,美國Welchrom公司);HLB固相萃取柱(美國Waters公司);其余試劑均為分析純。
1.2 標準溶液的配制
標準儲備溶液(1.0 mg/mL):準確稱取適量培氟沙星、氧氟沙星、諾氟沙星、洛美沙星及其相應的同位素內標物標準品,用甲醇分別配制成1.0 mg/mL的單標儲備溶液,置于-20 ℃下避光儲存。標準中間溶液(10.0 mg/L):分別移取0.1 mL標準儲備溶液于10 mL容量瓶中,用甲醇分別稀釋成質量濃度為10.0 mg/L的單標中間溶液,置于-20 ℃下避光儲存。
混合標準中間溶液(1.0 mg/L):取上述標準中間溶液各1 mL于10 mL容量瓶中,用甲醇稀釋成質量濃度為1.0 mg/L的混合標準中間溶液,置于-20 ℃下避光儲存;取上述同位素內標物的標準中間溶液各1 mL于10 mL容量瓶中,用甲醇稀釋成質量濃度為1.0 mg/L的同位素內標物混合標準中間溶液,置于-20 ℃下避光儲存。混合標準工作溶液(100.0 μg/L):取上述混合標準中間溶液5 mL于50 mL容量瓶中,用甲醇稀釋成質量濃度為100.0 μg/L的混合標準工作溶液,置于4 ℃下避光儲存;取上述同位素內標物的混合標準中間溶液5 mL于50 mL容量瓶中,用甲醇稀釋成質量濃度為100.0 μg/L的同位素內標物混合標準工作溶液,置于4 ℃ 下避光儲存。
1.3 樣品制備
實驗所用牛毛采自當地肉牛養殖場,培氟沙星、氧氟沙星、諾氟沙星、洛美沙星檢測結果均為陰性。
毛發的清洗:使用濃度1%SDS溶液浸泡毛發30 min,隨后用水漂洗多次至漂洗液澄清,置于40 ℃恒溫烘箱1 h后,取出晾干,剪碎成1~2 mm,存放于干燥器中,備用。
1.4 樣品前處理
準確稱取50 mg經上述處理后的毛發樣品于15 mL離心管中,滴加濃度為100.0 μg/L的混合內標溶液20 μL和5 mL 1%乙酸-乙腈溶液。室溫下超聲反應30 min,于5 000 r/min離心5 min,取上層清液至事先已加入100 mg PSA和100 mg C18吸附劑的15 mL離心管中,渦旋30 s后于5 000 r/min離心5 min,將上清液轉移到另一離心管中,在45 ℃條件下氮氣吹至近干,加入1 mL流動相(0.1%甲酸水溶液)溶解,使用0.22 μm有機濾膜過濾后,待上機檢測。
1.5 色譜條件
色譜柱:ZORBAX Eclipse Plus C18柱(100 mm×2.1 mm,1.8 μm)(Agilent);流動相:A為0.1%甲酸水溶液,B為甲醇;梯度洗脫程序為:0~1 min,B保持10%;1~5 min,B由10%線性升至50%;5~6 min,B由50%線性降至10%;6~6.5 min,B保持10%。流速:0.3 mL/min;進樣量:5 μL;柱溫:35 ℃。
1.6 質譜條件
電離模式:電噴霧電離正離子(ESI+)模式;檢測方式:多反應監測(MRM)模式;毛細管電壓:3 000 V;離子源溫度:150 ℃;干燥氣溫度:150 ℃;干燥氣流量:15 L/min;霧化氣壓力:30 psi;鞘氣溫度:300 ℃;鞘氣流量:11 L/min。
2 結果與討論
2.1 超高效液相色譜-串聯質譜分析結果
隨著超高效液相色譜-串聯質譜法在藥物殘留檢測的廣泛應用,使得分析檢測更高效靈敏,尤其適合大批量樣品的分析檢測[21]。本研究采用超高效液相色譜,以0.1%甲酸水溶液-甲醇做流動相,分離4種違禁氟喹諾酮類獸藥,通過質譜定性及定量檢測。根據氟喹諾酮類化學結構特點,容易得到H+而形成[M+H]+準分子離子,故本研究選擇正離子模式檢測,建立畜禽毛發中4種氟喹諾酮類藥物殘留的定性定量檢測方法。該方法分離效果較好,結果見圖1。
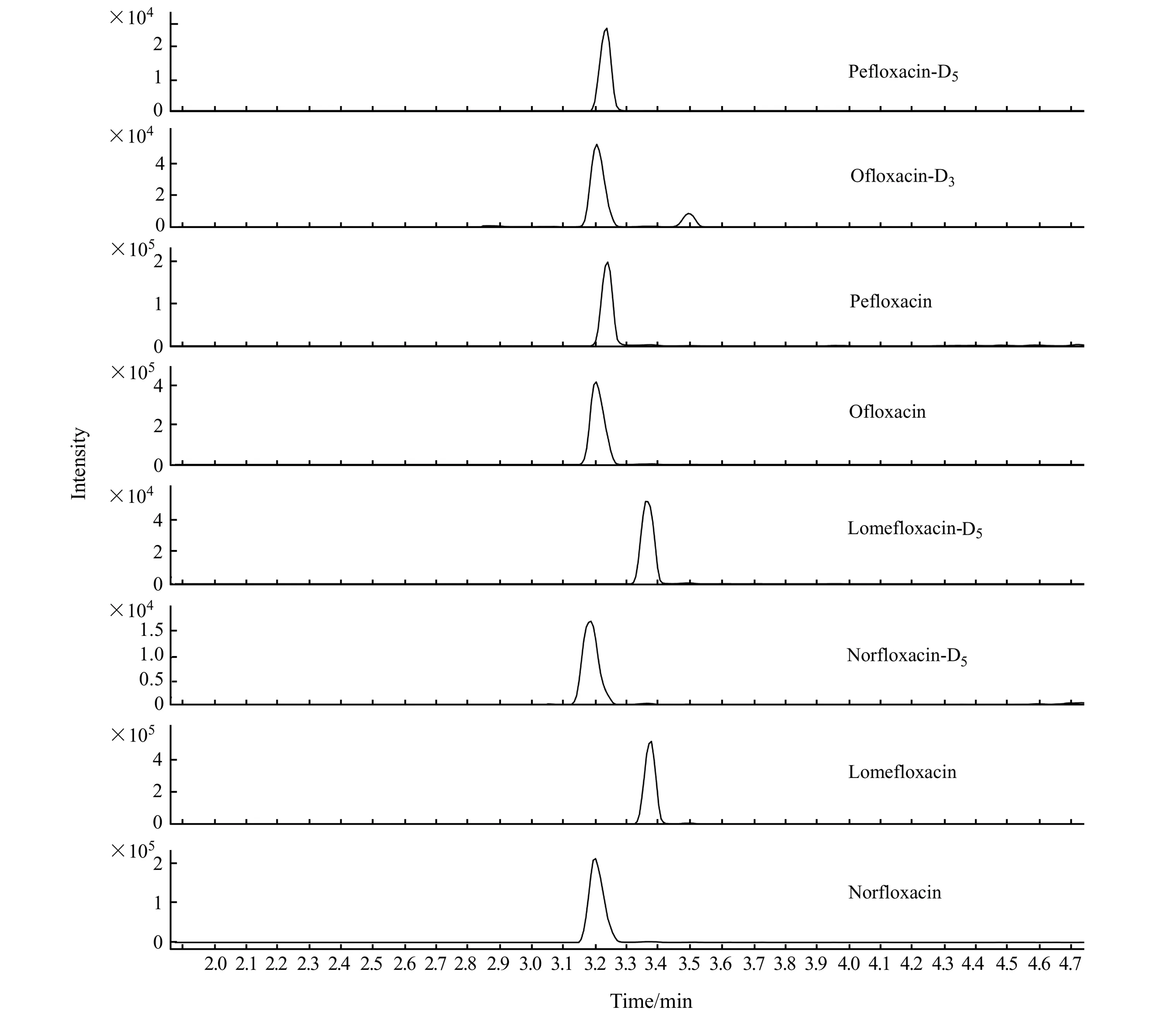
圖1 培氟沙星、氧氟沙星、諾氟沙星、洛美沙星及其內標物標準的定量離子色譜圖Fig.1 Chromatograms of quantitative ion for pefloxacin,ofloxacin,norfloxacin,lomefloxacin and their internal standards
采用內標法對樣品進行定量分析,在一定程度上可消除因為操作條件和檢測儀器帶來的誤差,進一步提高檢測結果的準確度,適用于畜禽毛發樣品中培氟沙星、氧氟沙星、諾氟沙星、洛美沙星這4種違禁氟喹諾酮類獸藥殘留的痕量分析檢測。檢測參數見表1。
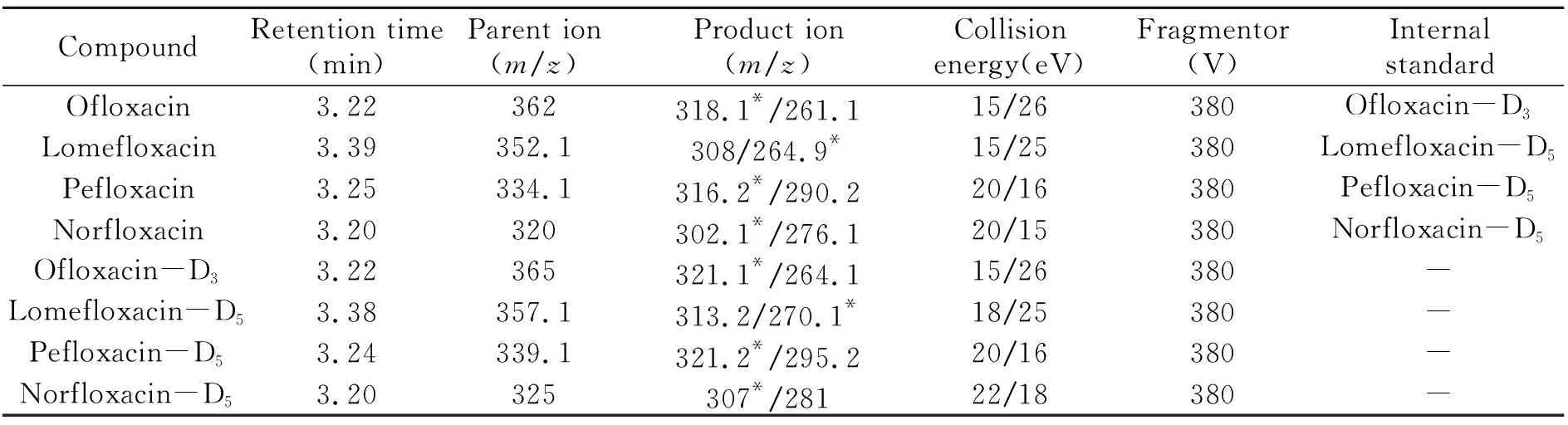
表1 目標化合物及其內標檢測參數Table 1 Detection parameter of target compounds and the internal standard
2.2 前處理方法的優化
2.2.1 提取方法優化畜禽毛發結構致密,藥物通常殘留于毛髓質中,外部有較厚的角質層包裹,因此需采用剪碎研磨的處理方式,使毛發的角質層破碎,毛髓質中藥物呈游離態進入提取溶液,通過水解的處理方式破壞毛發的內部結構,使藥物從中釋放出來。依據畜禽毛發樣品的吸附特性和氟喹諾酮類藥物的溶解性,對毛發中培氟沙星、氧氟沙星、諾氟沙星、洛美沙星的提取溶劑進行了優化。分別在毛發樣品中加入2%甲酸甲醇溶液、乙腈-50%HCl(1∶1,V/V)、乙腈-1%HAc溶液、乙腈-2%甲酸溶液、乙腈-磷酸鹽溶液,并添加50 μL 1 mg/L混合標準品溶液,其余步驟按“1.4”處理,研究不同提取溶劑的加標回收率。結果如圖2所示,經乙腈-1%HAc溶液超聲提取后樣品色譜峰雜質干擾最小,可以最大程度地去除毛發的基質成分,其回收率均高于其他4種提取溶劑,因而將乙腈-1%HAc溶液作為本實驗的提取溶劑。
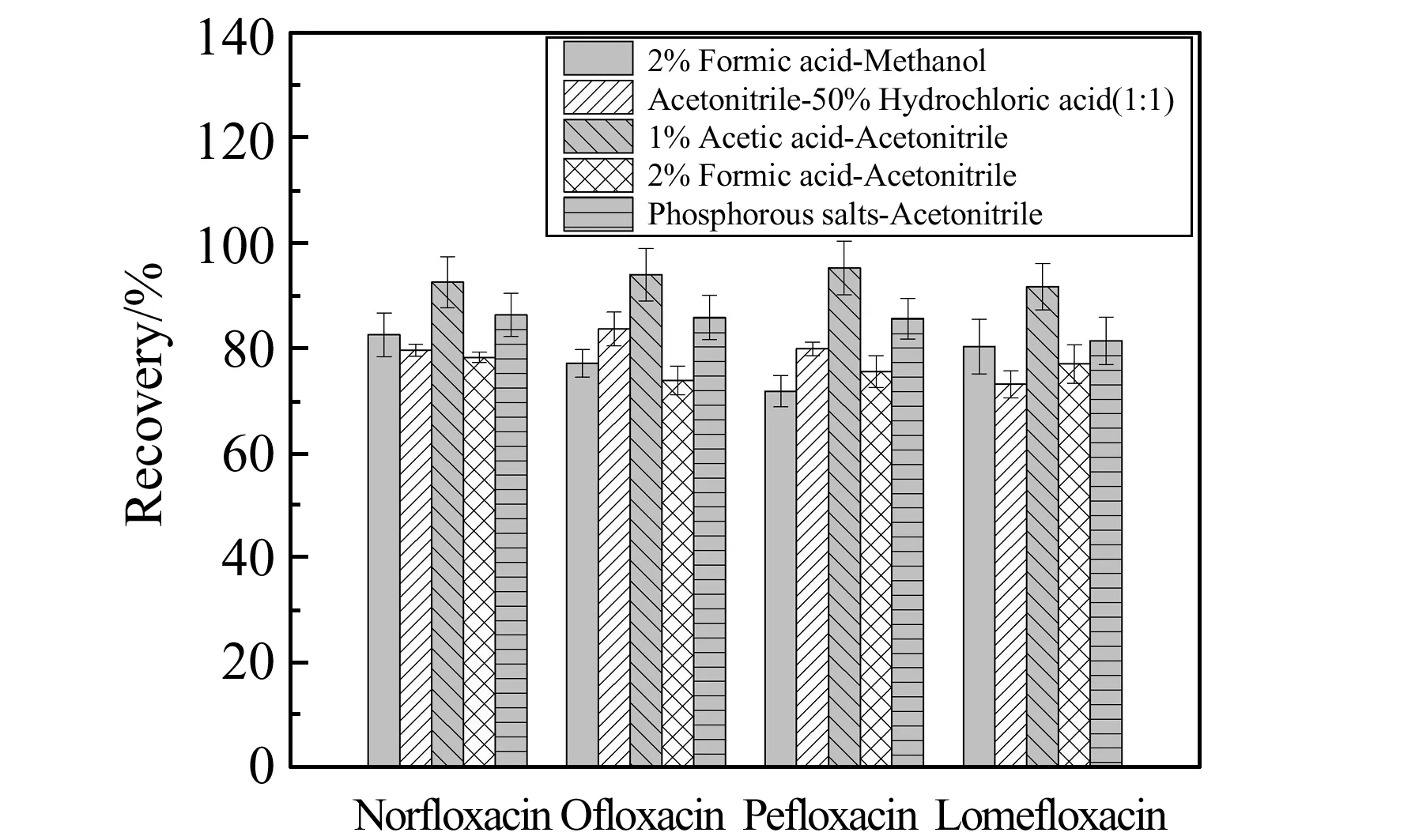
圖2 不同提取溶劑的回收率Fig.2 Recovery of different extraction solvents
對比分析了不同水浴溫度和超聲振蕩時間對培氟沙星、氧氟沙星、諾氟沙星、洛美沙星的提取效果,篩選出3組較優的方案進行進一步優化。制備了添加50 μL 1 mg/L混合標準品溶液的樣品,用乙腈-1%HAc溶液提取后,分別經25 ℃水浴超聲振蕩2 h、65 ℃水浴超聲振蕩2 h、80 ℃水浴超聲振蕩1 h處理,然后在5 000 r/min下離心5 min,取200 μL上清液,再加入800 μL初始流動相(0.1%甲酸甲醇溶液)混勻,濾膜過濾后上機檢測。3種不同水解條件下4種目標物的回收率如圖3所示,水浴溫度25 ℃和65 ℃的提取回收率高于80 ℃提取條件,可見升高水浴溫度后目標化合物的提取效果不佳。考慮實驗操作條件的簡便和節能因素,最終選擇25 ℃條件下水浴超聲振蕩2 h。
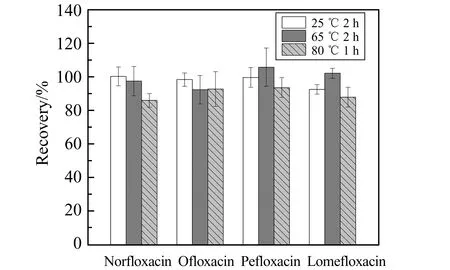
圖3 不同水解方法的回收率Fig.3 Recovery of different hydrolysis methods
2.2.2 凈化方法優化水解反應后的毛發產物一般要進行提取及凈化處理,以此來降低干擾物對檢測的影響,并達到濃縮分析物的目的。通常采用液-液提取(LLE)和固相萃取(SPE)的方式完成提取和凈化步驟。其中,SPE多數使用NH2、SCX、C18、GCB及混合模式(C18+SCX)等[22,23]。本方法選擇PSA/C18凈化管和HLB固相萃取柱分別對毛發提取液進行凈化,對比兩種方法的加標回收率。結果顯示,經PSA/C18凈化管處理的樣品回收率和凈化效果較好,因此選用PSA/C18凈化管進行毛發提取液凈化。
2.3 基質效應
本文選擇經樣品前處理后的空白基質和純溶劑分別稀釋標準品,配制標準曲線,用二者的斜率計算基質效應,即:(空白基質液標準曲線斜率/純溶劑標準曲線斜率-1)×100%,該值為負數表示基質對化合物的響應有抑制作用,該值為正數表示基質對化合物的響應有增強作用,其絕對值越大抑制或增強的作用越強。實驗結果表明,基質效應對4種化合物均為抑制作用,但影響不大,具體見表2。
2.4 線性范圍、檢出限和定量限
采用空白基質溶液配制質量濃度為0.2、0.5、1.0、2.0、5.0、10.0、20.0 μg/L的系列標準溶液,其中同位素內標濃度為2 μg/L,標準曲線方程及相關系數結果見表2。4種目標化合物在0.2~20 μg/L范圍內成線性相關,相關系數r2均大于0.993,線性良好,將標準溶液稀釋至信噪比(S/N)為3和10時的濃度分別定為目標化合物的檢測限和定量限,結果得出氧氟沙星和諾氟沙星的檢測限為0.05 μg/kg,定量限為0.1 μg/kg;洛美沙星和培氟沙星的檢測限為0.08 μg/kg,定量限為0.2 μg/kg(表2)。
2.5 回收率和精密度
參照方法定量限,在陰性樣品中添加了由低到高3個濃度水平的標準品混合溶液,每個濃度做6組平行實驗,分別計算加標回收率及其相對標準偏差(RSD),結果見表2。本方法回收率在87.09%~114.95%范圍內,RSD在0.13%~4.92%之間。Xiang等[24]采用超高效液相色譜-串聯質譜檢測雞毛中的諾氟沙星殘留,方法檢出限為0.3 g/kg,定量限為1.0 g/kg,回收率為89.2%~95.7%,RSD均小于10%。本實驗結果與其結果相似,方法滿足畜禽毛發中氟喹諾酮類藥物的分析檢測。
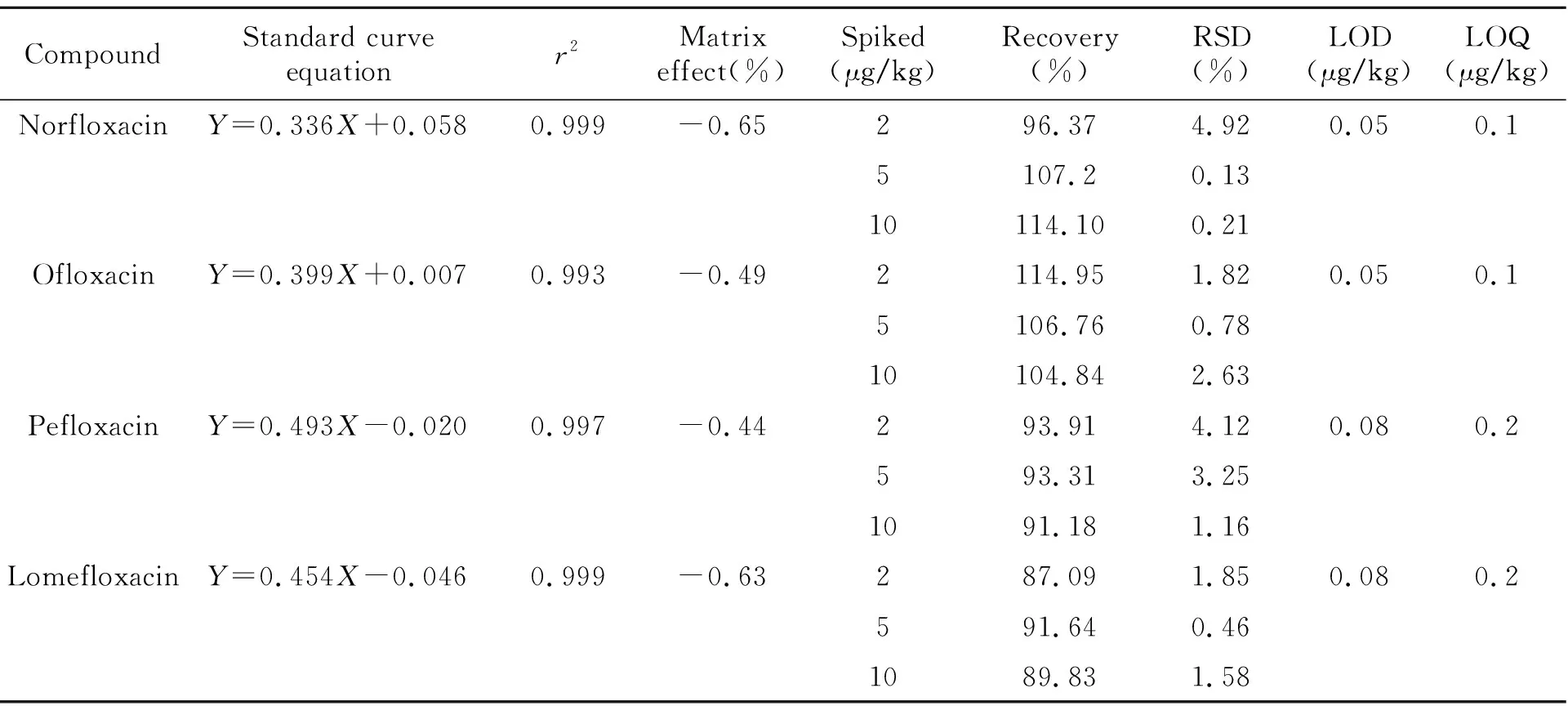
表2 標準曲線,基質效應,回收率,檢出限(LOD)和定量限(LOQ)Table 2 Standard curve equation,matrix effect,recovery,LOD and LOQ
2.6 實際樣品檢測
應用所建立的方法,在農貿市場共采集9份麻雞毛發樣品和3份肉牛毛發樣品進行檢測。檢出2份麻雞毛發中殘留氧氟沙星,其殘留水平為0.2651 μg/kg和0.2768 μg/kg。2份麻雞毛發陽性樣品檢測色譜圖見圖4,檢出氧氟沙星的陽性樣品色譜峰形良好。

圖4 實際樣品色譜圖Fig.4 Chromatogram of actual sample
3份肉牛毛發樣品均未發現存在培氟沙星、氧氟沙星、諾氟沙星、洛美沙星殘留。說明毛發可作為畜禽使用氟喹諾酮類藥物的檢測基質,適用于畜禽養殖的全過程監管。由此可見,上述所建立的方法具有很強的實際應用價值。
3 結論
利用超高效液相色譜-串聯質譜聯用技術,建立了一種利用畜禽毛發檢測培氟沙星、氧氟沙星、諾氟沙星和洛美沙星4種違禁氟喹諾酮類獸藥的分析方法。該方法前處理簡單,檢測限及定量限較低,有效地提供了一種借助畜禽毛發來監管養殖過程中違禁藥物培氟沙星、氧氟沙星、諾氟沙星、洛美沙星等違規使用的新思路,為畜禽產品質量安全監管提供了新的技術手段。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
