木質(zhì)素衍生酚類模化物原位加氫脫氧制取苯的研究
2021-09-16 08:15:12謝嘉琪駱仲泱李思敏孫浩然
能源工程 2021年4期
關(guān)鍵詞:催化劑實(shí)驗(yàn)
謝嘉琪,駱仲泱,李思敏,薛 爽,孫浩然
(浙江大學(xué) 能源清潔利用國(guó)家重點(diǎn)實(shí)驗(yàn)室,浙江 杭州310027)
0 引 言
對(duì)于木質(zhì)纖維素類生物質(zhì)中的纖維素和半纖維素的研究已經(jīng)比較成熟,可以取得較好的轉(zhuǎn)化效果。但是,對(duì)于占比接近1/3的木質(zhì)素卻難以物盡其用[1-5]。木質(zhì)素解聚后會(huì)生成各種酚類物質(zhì)[6,7],酚類含有天然的芳環(huán)結(jié)構(gòu),可以直接通過加氫脫氧過程轉(zhuǎn)化為芳香烴如BTX、環(huán)烷烴類化合物及其衍生物,在生產(chǎn)平臺(tái)化合物或高品位液體燃料方面存在巨大潛力[8,9]。
常規(guī)的加氫脫氧反應(yīng)(HDO)需要在高溫下進(jìn)行并提供高氫氣分壓,存在諸多問題:(1)需要額外提供大量的氫氣,有一定危險(xiǎn)性且成本高;(2)反應(yīng)過程中的高壓對(duì)設(shè)備要求高,且裝置難以實(shí)現(xiàn)小型化,提高了前期投入成本;(3)催化劑容易積碳失活[10-17]。近年來,對(duì)于木質(zhì)素衍生酚類的HDO條件逐漸趨于溫和,從一開始需要外加高壓氫氣,到后面臨氫反應(yīng),再到部分研究中使用供氫溶劑來提供HDO所需要的氫氣。目前,有部分催化劑在常壓下加氫效果較好,并且也有關(guān)于反應(yīng)時(shí)不提供氫氣而使用醇進(jìn)行原位供氫的探究[18,19]。
Song[20]等在探究木質(zhì)素在醇體系中,通過Ni/C催化劑的解聚機(jī)理時(shí)發(fā)現(xiàn),反應(yīng)時(shí)是否通入氫氣對(duì)于木質(zhì)素的轉(zhuǎn)化無(wú)影響,猜測(cè)醇可以提供活性氫物質(zhì)。隨后,通過同位素(H/D)標(biāo)記實(shí)驗(yàn)進(jìn)行進(jìn)一步驗(yàn)證。實(shí)驗(yàn)結(jié)果證實(shí),醇可以在鎳催化劑上分解產(chǎn)生氫氣并提供給木質(zhì)素進(jìn)行解聚反應(yīng)。Klein[1]等在Song的實(shí)驗(yàn)基礎(chǔ)上進(jìn)一步探索Ni/C催化劑的金屬負(fù)載量對(duì)反應(yīng)的影響。結(jié)果發(fā)現(xiàn),隨金屬負(fù)載量增加,反應(yīng)結(jié)束后容器中的壓力增大,且產(chǎn)物中不飽和鍵減少,由此推測(cè)加入催化劑中Ni的負(fù)載量越高,反應(yīng)中生成的H2越多。Wang[21]等將苯酚和異丙醇混合后,在催化劑Raney Ni和β沸石的催化下反應(yīng)4 h(433K),可以將苯酚轉(zhuǎn)化為大量的苯。其中異丙醇作為溶劑和氫源,水相重整制氫以供給苯酚氫解反應(yīng)。但是,異丙醇成本太高,每噸5000~6000元不等,而純苯的一噸價(jià)格不到4000元,毫無(wú)性價(jià)比可言;并且雷尼鎳不能離開乙醇或水,即必須保持濕潤(rùn),否則馬上自燃,同時(shí)也容易失去活性。Lin[22]等采用催化劑Pt/α-MoC催化甲醇進(jìn)行水相重整制氫,測(cè)得反應(yīng)的產(chǎn)氫速率極高,且碳收率高。Hu[23]等探究了在原位供氫體系下木質(zhì)素的氫解。實(shí)驗(yàn)以異丙醇為供氫體,使用PtRe/TiO2作為催化劑催化木質(zhì)素在240℃下反應(yīng)12 h,獲得了18.71%的單酚。解聚過程中β-O-4鍵被完全破壞,解聚效果良好。實(shí)驗(yàn)發(fā)現(xiàn):在外加氫氣的情況下,單酚產(chǎn)率有所下降,推測(cè)是競(jìng)爭(zhēng)吸附發(fā)生在了催化劑表面活性位上。
使用甲醇等溶劑作為氫源取代常規(guī)的外加高壓氫氣,操作更加安全;另外,反應(yīng)在常壓下進(jìn)行,設(shè)備簡(jiǎn)單,安全性高。
本文探究了在原位供氫體系下,使用Ru/C催化劑,采用三種典型木質(zhì)素衍生酚類模化物苯酚、愈創(chuàng)木酚和2,6-二甲氧基苯酚,分別考察了催化劑的金屬負(fù)載量、反應(yīng)時(shí)間、甲醇質(zhì)量等反應(yīng)條件對(duì)加氫脫氧效果的影響,通過對(duì)反應(yīng)條件的調(diào)控,可以實(shí)現(xiàn)溫和條件下苯的較高收率。
1 材料與方法
1.1 實(shí)驗(yàn)材料及催化劑制備
本實(shí)驗(yàn)使用的苯酚和三水氯化釕(RuCl3·3H2O)為阿拉丁公司生產(chǎn),愈創(chuàng)木酚及2,6-二甲氧基苯酚均為Sigma-Aldrich公司的產(chǎn)品,無(wú)水甲醇和活性炭為國(guó)藥公司生產(chǎn),5%Ru/C催化劑為TCI生產(chǎn)的商用催化劑。
實(shí)驗(yàn)所使用的1%Ru/C和3%Ru/C催化劑采用等體積浸漬法制備得到。根據(jù)實(shí)驗(yàn)所需的不同負(fù)載量,先將RuCl3·3H2O溶于去離子水中,配置成不同濃度的均勻溶液;然后稱取定量的活性炭,將RuCl3溶液少量多次緩慢滴入活性炭中,攪拌均勻,重復(fù)在負(fù)壓下超聲波處理30 min,放入烘箱中80℃烘干過夜。次日取出,在10%H2/Ar氣氛中、400℃對(duì)催化劑進(jìn)行焙燒還原3 h,最后在0.5%O2/N2氣氛下進(jìn)行鈍化處理。
1.2 催化劑表征
采用XRD對(duì)催化劑表面的金屬分布進(jìn)行分析。使用的儀器是X'Pert PRO,采用單色Cu-Kα輻射(入射角=0.5°,步長(zhǎng)=0.026°,積分時(shí)間24 s/step),在40 mA和40 k V下工作。
為了進(jìn)一步研究金屬Ru的負(fù)載狀態(tài),采用XPS(X射線光電子能譜)對(duì)Ru的存在和價(jià)態(tài)進(jìn)行表征。使用Thermo Scientific K-Alpha+光譜儀,通過能量100 eV進(jìn)行全光譜掃描,通過能量30 eV用于窄光譜掃描。以284.8 eV處的C1s峰為結(jié)合能的參考,以糾正由電荷效應(yīng)引起的位移。
使用Micromeritics公司的TRISTAR 3020對(duì)催化劑進(jìn)行氮?dú)馕摳椒治觯肂ET方法計(jì)算催化劑比表面積,采用t-Plot計(jì)算微孔面積;催化劑的介孔分布和平均孔徑分布通過BJH(Barret-Joyner-Hallenda)方法計(jì)算N2吸附支可得到。
1.3 實(shí)驗(yàn)方法
本實(shí)驗(yàn)為木質(zhì)素酚類模化物在甲醇水相重整制氫體系中直接原位進(jìn)行的加氫反應(yīng)。提前將實(shí)驗(yàn)所需Ru/C催化劑在10%H2/Ar氣氛中400℃還原2 h。實(shí)驗(yàn)時(shí),先將反應(yīng)原料加入高壓反應(yīng)釜釜體中,裝好反應(yīng)釜后,反復(fù)通入N2三次進(jìn)行排空,設(shè)置好反應(yīng)溫度(230℃)、時(shí)間(4~16 h)、轉(zhuǎn)速(600 r/min)等參數(shù)后進(jìn)行反應(yīng)。
1.4 催化劑穩(wěn)定性測(cè)試
反應(yīng)完成后,通過離心及過濾固液混合物來回收催化劑(固體)。回收的濕催化劑通過丙酮和乙醇清洗后110℃烘干12 h,以除去濕催化劑的殘留溶劑,再用于下一輪實(shí)驗(yàn)。循環(huán)使用時(shí)保持S/C比(底物與催化劑的摩爾比)恒定。
1.5 產(chǎn)物分析
待反應(yīng)釜的溫度降至室溫后,氣體首先通過排氣閥進(jìn)行收集,這部分氣體之后通過GC-FID/TCD對(duì)其進(jìn)行定性分析。再打開反應(yīng)釜以收集液體,并用乙酸乙酯進(jìn)行多次萃取操作。萃取后,將液體離心分離并通過0.22μm注射式過濾器過濾以分離出固體殘留物。對(duì)液體產(chǎn)物采用GC-MS進(jìn)行定性分析。實(shí)驗(yàn)使用的GC-MS(Agilent-7890 GC和Agilent MS-5977A MSD)配備DB-WAX MS色譜柱(0.25μm×0.25 mm×30 m),使用的升溫程序與下面GC-FID的升溫程序一致,使用NIST庫(kù)用于譜圖識(shí)別。液體產(chǎn)物通過GC-FID進(jìn)行定量分析。GC配備HP-5毛細(xì)管柱和火焰離子化(FID)檢測(cè)器,柱箱升溫程序如下:在40℃停留3 min,然后升至180℃(7℃/min),再升至到280℃(10℃/min),停留10 min。所使用的噴射器溫度為270℃,檢測(cè)器保持在300℃。
對(duì)使用后的催化劑在PerkinElmer Pyris Diamond差示掃描量熱儀上進(jìn)行熱重-差熱分析(TG-DTA)。在熱重實(shí)驗(yàn)前,回收催化劑用丙酮和乙醇洗滌烘干后,將樣品在通入空氣(20 mL/min)的情況下,以5℃/min的加熱速率從室溫升至900℃,并記錄下質(zhì)量隨溫度變化的曲線。
分別用以下公式計(jì)算轉(zhuǎn)化率(Conversion),選擇性(Selectivity)和產(chǎn)率(Yield),未反應(yīng)的部分不包括在產(chǎn)物選擇性的計(jì)算中,其中的n代表反應(yīng)物或產(chǎn)物的摩爾數(shù)。

2 結(jié)果與討論
2.1 催化劑表征
實(shí)驗(yàn)使用的催化劑在反應(yīng)前均進(jìn)行了以下各項(xiàng)表征測(cè)試。
圖1為不同金屬負(fù)載量的Ru/C催化劑的XRD結(jié)果譜圖,把其中30°到42°范圍內(nèi)的貴金屬特征峰單獨(dú)截取出來放大。可以看出,曲線比較平滑,無(wú)明顯金屬釕的特征衍射峰,說明負(fù)載金屬分布均勻,分散性良好。
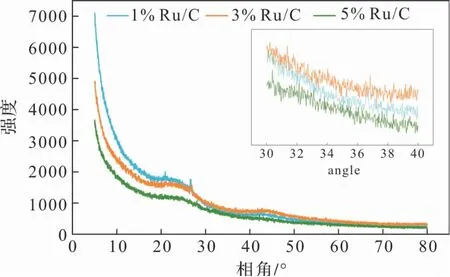
圖1 催化劑的XRD譜圖及30°到42°貴金屬特征峰放大圖(嵌入)
圖2 為各催化劑的XPS譜圖。從圖中可以看出,電子結(jié)合能為462 eV的位置代表的是Ru的3p3/2軌道特征峰,電子結(jié)合能為485 eV的位置對(duì)應(yīng)的是Ru的3p1/2軌道特征峰,這兩個(gè)峰對(duì)應(yīng)的是0價(jià)態(tài)Ru的電子結(jié)合能[24],因此可以推斷出活性炭上面負(fù)載的金屬Ru是以0價(jià)態(tài)即金屬單質(zhì)形式均勻分布在載體表面。
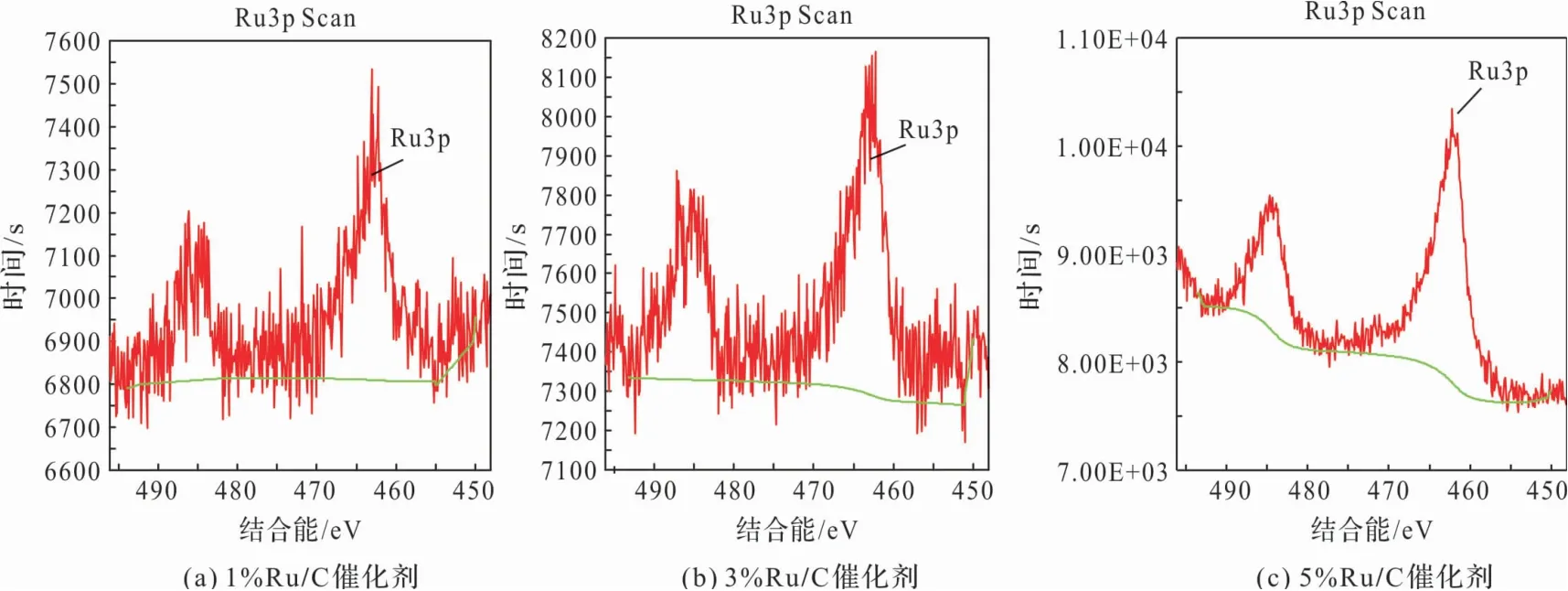
圖2 各催化劑的XPS譜圖
表1為各催化劑的BET數(shù)據(jù),孔道特性均通過BET測(cè)試方法進(jìn)行計(jì)算分析。結(jié)果表明,在金屬負(fù)載量最大為5%時(shí),催化劑比表面積最小,僅為754 m2/g;而金屬負(fù)載量最小的1%Ru/C擁有最大的比表面積(1306 m2/g);比表面積和微孔面積隨著負(fù)載量增加而逐漸減小,平均孔徑有一定增加,推測(cè)是負(fù)載量增加,部分微孔孔道被負(fù)載的金屬堵塞。

表1 BET表征數(shù)據(jù)
2.2 加氫脫氧實(shí)驗(yàn)結(jié)果分析
本文使用原位供氫體系對(duì)不同酚類模化物進(jìn)行HDO實(shí)驗(yàn)。
2.2.1 催化劑金屬負(fù)載量的影響
選用最簡(jiǎn)單的苯酚做酚類模化物,在這個(gè)過程中進(jìn)行催化劑篩選,實(shí)驗(yàn)結(jié)果如圖3所示。液體產(chǎn)物主要有苯、甲苯、環(huán)己醇和環(huán)己烷,且液體產(chǎn)物的摩爾平衡均在90%以上。從圖3可以看出,苯酚的轉(zhuǎn)化率在金屬負(fù)載量增加的情況下,有顯著提高,各產(chǎn)物的收率也明顯提升。具體來說,使用1%Ru/C催化劑時(shí),苯酚的轉(zhuǎn)化率為32%;同時(shí),苯的收率僅為10%,沒有檢測(cè)到環(huán)己烷,環(huán)己醇的收率也僅為11%。隨著金屬負(fù)載量的增加,5%Ru/C作用下的苯酚幾乎全部轉(zhuǎn)化,轉(zhuǎn)化率接近100%;同時(shí),苯的產(chǎn)率提升至60%,環(huán)己烷的產(chǎn)率也增加至27%,HDO效果提升顯著。推測(cè)原因是:由于負(fù)載量增加后,增多的貴金屬引入了更多的活性位點(diǎn)。對(duì)于本實(shí)驗(yàn)體系而言,甲醇水相重整制氫以及酚類HDO均需在金屬活性位點(diǎn)上進(jìn)行,因此,較高的金屬負(fù)載量和較好的分散程度能夠更好地促進(jìn)反應(yīng)向更加完全的方向進(jìn)行。

圖3 不同金屬負(fù)載量的催化劑的反應(yīng)產(chǎn)物分布;反應(yīng)工況:苯酚(0.2 g),催化劑(0.1 g),230℃,1 atm N2,醇油比5∶1,溶劑(12 mL),12 h
對(duì)反應(yīng)后收集到的氣體采用GC-FID/TCD對(duì)其進(jìn)行定性分析。圖4為苯酚進(jìn)行加氫脫氧實(shí)驗(yàn)后氣體的檢測(cè)結(jié)果,可以看出,氣體中存在36%的氫氣,證明了本體系確實(shí)存在甲醇水相重整制氫反應(yīng)。

圖4 氣體產(chǎn)物GC-FID/TCD檢測(cè)譜圖
2.2.2 反應(yīng)時(shí)間的影響
根據(jù)前面的催化劑篩選結(jié)果,后續(xù)實(shí)驗(yàn)均統(tǒng)一采用5%Ru/C催化劑,催化不同酚類模化物(苯酚、含有甲氧基的愈創(chuàng)木酚和2,6-二甲氧基苯酚)進(jìn)行加氫實(shí)驗(yàn)反應(yīng),以驗(yàn)證在本實(shí)驗(yàn)體系中酚類物質(zhì)的HDO反應(yīng)規(guī)律以及甲氧基官能團(tuán)的影響。采用醇油比為5∶1、反應(yīng)溫度230℃,不同反應(yīng)時(shí)間(4 h、8 h、12 h、16 h)的實(shí)驗(yàn)結(jié)果如圖5所示。反應(yīng)進(jìn)行到4 h時(shí),愈創(chuàng)木酚和二甲氧基苯酚的產(chǎn)物中檢測(cè)到少量苯酚;反應(yīng)進(jìn)行到12 h時(shí),三種原料基本上全部轉(zhuǎn)化。對(duì)于不同的單酚,從產(chǎn)生苯的先后順序上來說:2,6-二甲氧基苯酚>愈創(chuàng)木酚>苯酚,推斷在本體系的HDO過程中,甲氧基首先被脫掉,再進(jìn)一步生成部分甲醇[19,25,26],同時(shí)它的存在和轉(zhuǎn)化過程降低了酚類加氫的難度。可以看出,隨反應(yīng)時(shí)間的增加,環(huán)己醇、環(huán)己酮呈現(xiàn)逐漸減少的趨勢(shì),而苯和環(huán)己烷逐漸增加,且苯的收率大于環(huán)己烷。可以推測(cè),隨著甲醇水相重整制氫以及酚類HDO的同時(shí)進(jìn)行,一開始酚類先脫掉甲氧基;隨著H增多,酚類開始進(jìn)行加氫反應(yīng),生成環(huán)己醇和環(huán)己酮,環(huán)己醇生成環(huán)己烷或者苯[21],推測(cè)的反應(yīng)路徑圖如圖6所示。由于體系中的氫源較少,抑制了過度加氫,環(huán)己醇生成苯的反應(yīng)占主導(dǎo)。在反應(yīng)進(jìn)行到一定程度以后,苯酚較之其他單酚能夠生成更多的苯。推測(cè)原因是:對(duì)于苯酚來說,不含甲氧基,因此沒有甲氧基脫除后生成的那一部分甲醇,體系中的氫源較少,活性氫也相對(duì)較少,故最后能夠生成更多的苯。

圖5 苯酚(a)、愈創(chuàng)木酚(b)和二甲氧基苯酚(c)的產(chǎn)物分布;反應(yīng)工況:苯酚(0.2 g),催化劑(0.1 g),230℃,1 atm N2,醇油比5∶1,溶劑(12 mL)

圖6 推測(cè)的簡(jiǎn)化反應(yīng)路徑
2.2.3 醇油比的影響
在本實(shí)驗(yàn)體系中,甲醇是主要的氫源,給酚類物質(zhì)HDO提供活性氫原子,是反應(yīng)的關(guān)鍵影響因素。改變醇油比,探究在醇油比為2.5∶1、5∶1、10∶1下的加氫效果,實(shí)驗(yàn)結(jié)果如圖7所示。從圖7可以看出,苯酚和愈創(chuàng)木酚的規(guī)律類似;隨醇油比增加,酚類轉(zhuǎn)化率略微降低,苯的收率顯著減少。這說明過多的甲醇對(duì)酚類模化物的轉(zhuǎn)化率影響較小,但會(huì)促進(jìn)加氫反應(yīng),促使環(huán)己醇向環(huán)己烷轉(zhuǎn)化,從而抑制苯的生成。這可能是由于較多的甲醇生成較多氫氣和活性氫原子,形成競(jìng)爭(zhēng)吸附,造成產(chǎn)物過度加氫;同時(shí),較多游離氫使得生成的環(huán)己醇難以進(jìn)一步產(chǎn)生苯。對(duì)于2,6-二甲氧基苯酚來說,甲醇的量對(duì)其HDO沒有明顯的影響,而對(duì)于2,6-二甲氧基苯酚,甲醇的量對(duì)其HDO沒有明顯的影響,推測(cè)原因是由于一分子二甲氧基苯酚含兩分子甲氧基,而根據(jù)前面的實(shí)驗(yàn)結(jié)果推測(cè),脫掉的甲氧基也參與供氫,故體系中的氫源相對(duì)而言處于過量的狀態(tài),對(duì)甲醇的量不敏感。

圖7 不同醇油比下的苯酚(a)、愈創(chuàng)木酚(b)和二甲氧基苯酚(c)的產(chǎn)物分布;反應(yīng)工況:苯酚(0.2 g),催化劑(0.1 g),230℃,1 atm N2,溶劑(12 mL),12 h
2.3 催化劑穩(wěn)定性分析
催化劑循環(huán)使用的效果如圖8所示。可以看出,連續(xù)使用三次后,苯酚的轉(zhuǎn)化率無(wú)明顯變化,生成的苯逐漸減少,環(huán)己醇逐漸增多,催化劑性能有略微降低。推測(cè)原因是:反應(yīng)中生成少量積碳,導(dǎo)致比表面積減小;在這種情況下,Ru分散度降低,部分催化位點(diǎn)失活,環(huán)己醇進(jìn)一步反應(yīng)的速率降低,導(dǎo)致下一步反應(yīng)的產(chǎn)物苯減少。
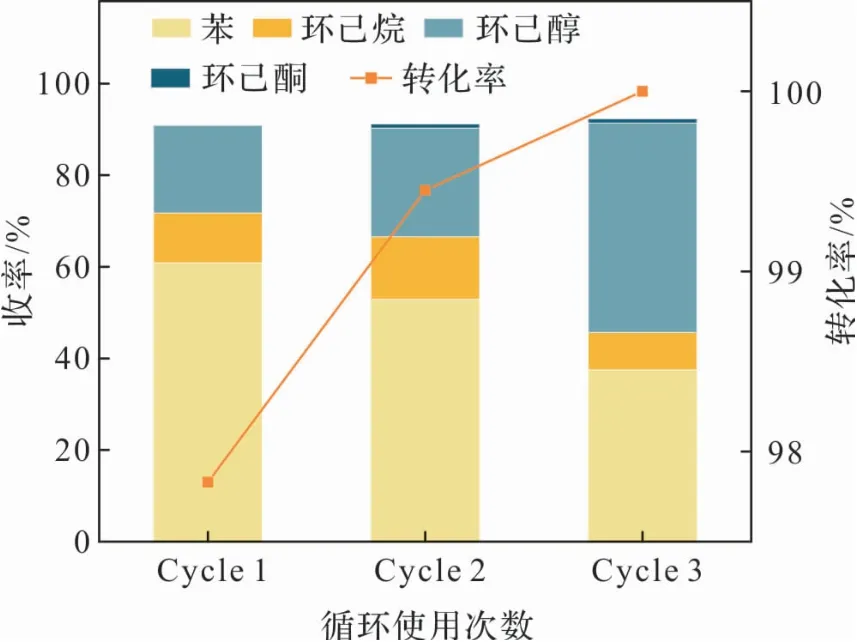
圖8 催化劑循環(huán)使用產(chǎn)物分布;反應(yīng)工況:苯酚(0.2 g),催化劑(0.1 g),230℃,1 atm N2,溶劑(12 mL),12 h
對(duì)使用前后的催化劑進(jìn)行熱重測(cè)試,以分析反應(yīng)后的積碳情況,測(cè)試結(jié)果如圖9所示。將新鮮的催化劑和使用后的催化劑做對(duì)比分析,可以認(rèn)為,兩者質(zhì)量差即為積碳量。可以看出,催化劑的積碳量較少,積碳情況較好,在400℃有一個(gè)額外的峰,推測(cè)為積碳峰[27]。

圖9 新鮮催化劑和使用后的催化劑的TG及DTG
3 結(jié) 論
(1)在本研究的原位供氫體系中,最優(yōu)工況下(常壓N2,反應(yīng)溫度230℃,醇油比2.5∶1,反應(yīng)時(shí)間12 h),三種酚類模化物均接近或達(dá)到100%轉(zhuǎn)化率,其中苯的收率最高可達(dá)76%。
(2)提出了可能的反應(yīng)路徑,即含甲氧基酚類模化物先脫掉甲氧基形成苯酚,隨后苯環(huán)加氫形成環(huán)己醇,再進(jìn)一步HDO產(chǎn)生脫氧產(chǎn)物。結(jié)合對(duì)醇油比影響的研究,發(fā)現(xiàn)從環(huán)己醇轉(zhuǎn)化苯和環(huán)己烷的路徑存在競(jìng)爭(zhēng),而體系中的活性氫原子數(shù)量對(duì)兩種脫氧產(chǎn)物的分布具有重要影響。
(3)由于含甲氧基的酚類模化物脫氧產(chǎn)物的產(chǎn)率相對(duì)較低,環(huán)己醇的產(chǎn)率較高,甲氧基對(duì)體系中的活性氫原子數(shù)量也具有一定貢獻(xiàn)。過多的活性氫原子會(huì)與反應(yīng)產(chǎn)物產(chǎn)生競(jìng)爭(zhēng)吸附,從而影響脫氧效果,同時(shí)也可能使已經(jīng)生成的苯進(jìn)一步過度加氫造成苯的選擇性降低。
(4)綜上所述,本研究在常壓氮?dú)庵校捎眉状甲鳛闅湓矗梢詫⒛举|(zhì)素衍生酚類化合物高效轉(zhuǎn)化為以苯為主的完全脫氧產(chǎn)物,實(shí)現(xiàn)了木質(zhì)素的安全、綠色轉(zhuǎn)化。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國(guó)資源綜合利用(2016年4期)2016-01-22 08:27:23
