復方參芪片的質量標準研究
2021-09-03 03:06:54崔曉博李明春程艷芹
藥學服務與研究 2021年4期
崔曉博,李明春,劉 旭,程艷芹
(海軍第九七一醫院藥劑科,山東青島 266071)
復方參芪片為海軍第九七一醫院的自制制劑[批準文號:總制字(2016)F405002],由補骨脂(君藥,具有補腎壯陽、固精縮尿、溫脾止瀉、納氣平喘等功效)、黃芪(君藥,具有益衛固表、利尿消腫、托毒生肌等功效)、丹參、降香等十五味藥材制成,具有疏風祛濕、通絡達表、滋補肝腎的功效,主治白癜風。該制劑經臨床使用多年,療效確切。為了更好地控制該制劑的質量,保證其臨床用藥的安全性和有效性,本研究對原質量標準中的薄層鑒別法進行修訂和增補,并增加了制劑中主要成分補骨脂的含量測定項。
1 儀器和試藥
1.1 儀器 JM-B2102型百分之一電子天平(余姚市紀銘稱重校驗設備有限公司);FA1604型萬分之一電子分析天平(上海衡平儀表廠);DV215CD型十萬分之一電子天平(美國OHAUS公司);SY2200-T型超聲波清洗器(上海聲源超聲儀器設備有限公司);Agilent 1260高效液相色譜儀(美國安捷倫公司);WD-9340C型紫外分析儀(北京六一儀器廠);電熱恒溫水浴鍋(上海梅香儀器有限公司)。
1.2 試藥 以羧甲基纖維素鈉為黏合劑的硅膠G薄層板(青島海洋化工廠分廠);補骨脂素(批號110739-201115,含量為99.9%)、異補骨脂素(批號110738-201313,含量為100%)、黃芪甲苷(批號110781-201314,含量為96.9%)、柴胡皂苷對照品(批號110778-201310,含量為97.3%),補骨脂(批號121056-200904)、丹參(批號120923-201414)、柴胡(批號120992-201108)、黃芪(批號121462-201304)、當歸(批號120927-201516)對照藥材均購于中國食品藥品檢定研究院;乙腈及甲醇均為一級色譜純(天津市四友生物醫學技術有限公司);復方參芪片樣品(批號20140421、20140910、20150317)、陰性樣品及蒸餾水均為海軍第九七一醫院自制;其他試劑均為分析純。
2 方法和結果
2.1 薄層鑒別
2.1.1 補骨脂的薄層鑒別[1-2]取復方參芪片,研細,稱取粉末10 g,加入甲醇40 ml,超聲20 min后過濾,濾液蒸干,殘渣加水20 ml溶解,用乙酸乙酯30 ml分兩次振搖提取,蒸干,殘渣加甲醇1 ml溶解,作為供試品溶液。取缺補骨脂的復方參芪片陰性樣品10 g,同法制成陰性對照溶液。取補骨脂對照藥材2 g,同法制成對照藥材溶液。取補骨脂素和異補骨脂素對照品,加入甲醇,制成濃度各為1 mg/ml的對照品溶液。吸取上述供試品溶液5 μl、對照藥材溶液7 μl、對照品溶液3 μl、陰性對照溶液5 μl,分別點于同一以羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以正己烷-乙酸乙酯(4∶1)為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視。供試品色譜中,在與對照藥材、對照品色譜相應的位置上,顯相同顏色的斑點(上層黃綠色斑點為異補骨脂素,下層淡黃色斑點為補骨脂素),而陰性對照溶液在相應位置上顯示藍色斑點,無干擾。見圖1。
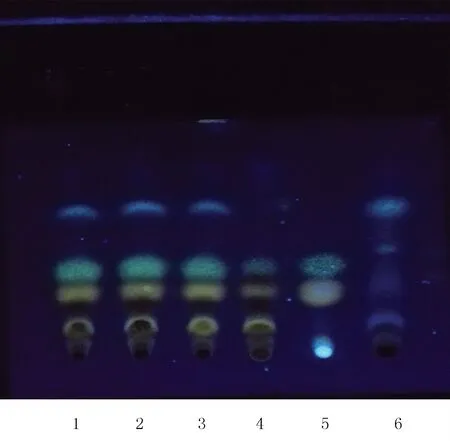
圖1 補骨脂的薄層色譜圖
2.1.2 丹參的薄層鑒別[2-3]取復方參芪片,研細,稱取粉末2 g,加水10 ml溶解,以相對離心力8.05×g離心5 min,取上清液,加稀鹽酸調pH至2,加乙酸乙酯20 ml,振搖提取,提取液置水浴蒸干,殘渣加甲醇1 ml溶解,作為供試品溶液。取缺丹參的復方參芪片陰性對照樣品2 g,同法制成陰性對照溶液。取丹參對照藥材2 g,同法制成對照藥材溶液。吸取上述3種溶液各5 μl,分別點于同一以羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以二氯甲烷-丙酮-甲酸(25∶10∶4)為展開劑,展開,取出,晾干后,置氨蒸汽中熏后,在空氣中放置10 min,置紫外光燈(365 nm)下檢視。供試品色譜中,在與對照藥材色譜相應的位置上顯黃綠色斑點,而缺丹參的復方參芪片陰性對照溶液在相應位置上顯藍色斑點,無干擾。見圖2。
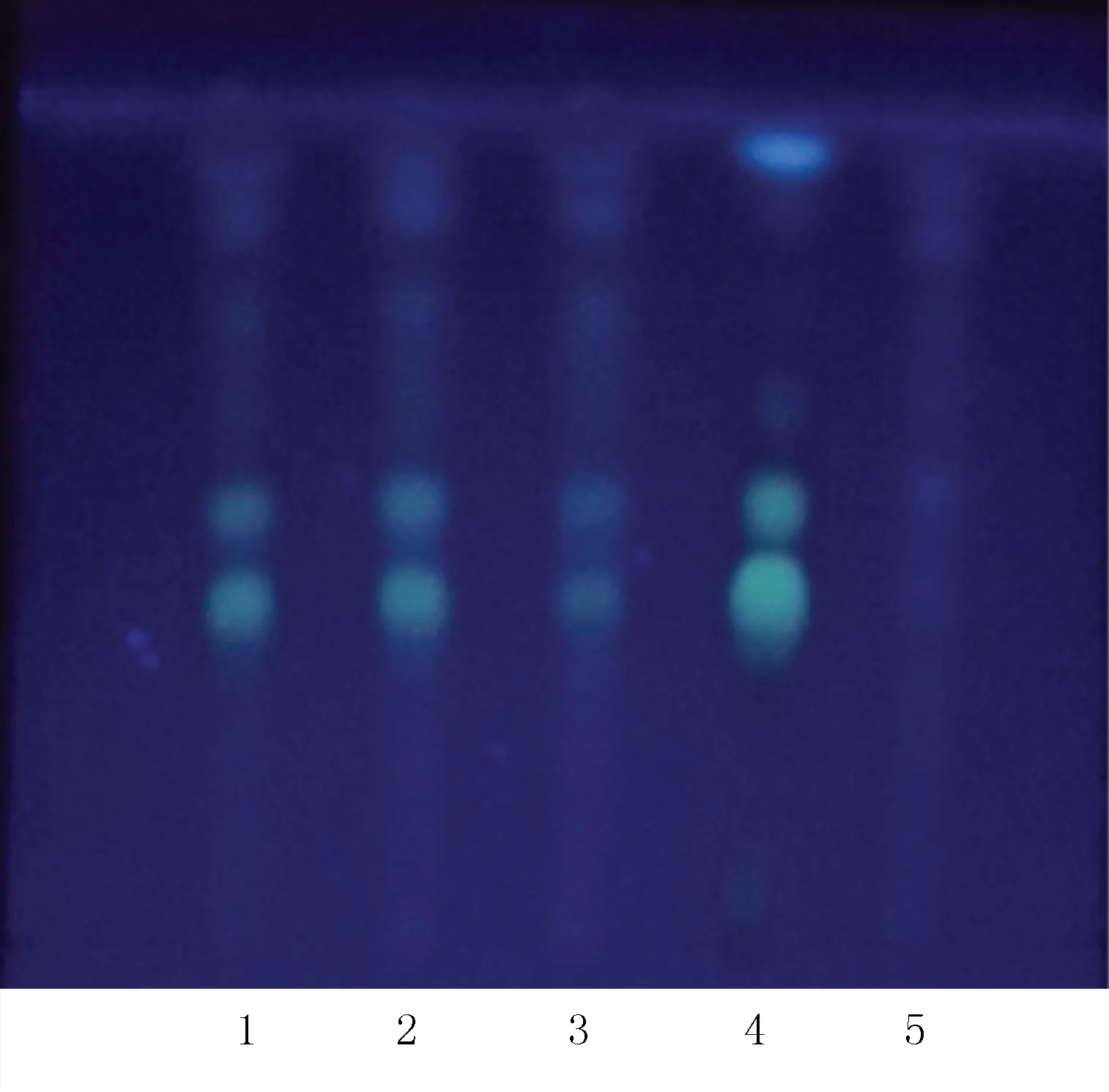
圖2 丹參的薄層色譜圖
2.1.3 柴胡的薄層鑒別[2,4]取復方參芪片,研細,稱取粉末10 g,加甲醇30 ml,超聲處理30 min,過濾,濾液蒸干,殘渣加水20 ml使溶解,加水飽和的正丁醇提取2 次(30 ml/次),合并正丁醇液,加氨試液(40 ml濃氨水加水稀釋到100 ml)40 ml洗滌,棄去氨試液,蒸干正丁醇,殘渣加甲醇1 ml溶解,作為供試品溶液。取缺柴胡的復方參芪片陰性對照樣品10 g,同法制成陰性對照溶液。取柴胡對照藥材1 g,加水50 ml,煮沸30 min,放冷,過濾,濾液加水飽和正丁醇振搖提取2次,同法制成對照藥材溶液。取柴胡皂苷對照品,加甲醇,制成濃度為1 mg/ml的對照品溶液。吸取復方參芪片供試品溶液5 μl、對照藥材溶液5 μl、對照品溶液5 μl、陰性對照溶液5 μl,分別點于同一以羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以正丁醇-乙酸乙酯-稀氨水(1→10,即將濃氨水體積稀釋10倍)(4∶1∶5)的上層溶液為展開劑[4],展開,取出,晾干,噴以2%對二甲氨基苯甲醛的硫酸乙醇溶液(1→10),105 ℃加熱至斑點顯色清晰。供試品色譜中,在與對照藥材和對照品色譜相應的位置上,均顯相同的粉紅色斑點,同時陰性對照溶液無干擾。見圖3。

圖3 柴胡的薄層色譜圖
2.1.4 黃芪的薄層鑒別[5-6]取復方參芪片,研細,稱取粉末10 g,加水飽和正丁醇60 ml,加熱回流1 h,濾過,濾液用1% NaOH溶液洗滌3次,每次15 ml,棄去NaOH溶液,用水飽和的正丁醇洗至中性,棄去水液,置水浴蒸干正丁醇,殘渣用甲醇1 ml溶解,作為供試品溶液。取缺黃芪的復方參芪片陰性對照樣品10 g,同法制成陰性對照溶液。另取黃芪對照藥材1 g,同法制成對照藥材溶液。取黃芪甲苷對照品,加甲醇制成濃度為1 mg/ml的對照品溶液。吸取上述4種溶液各3 μl,分別點于同一以羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以二氯甲烷-甲醇-水(65∶35∶10)的下層溶液為展開劑[5],展開,取出,晾干,噴以10%硫酸乙醇溶液,在105 ℃加熱至斑點顯色清晰。供試品色譜中,在與對照藥材色譜和對照品色譜相應的位置上,顯相同的棕色斑點,陰性對照無干擾。見圖4。
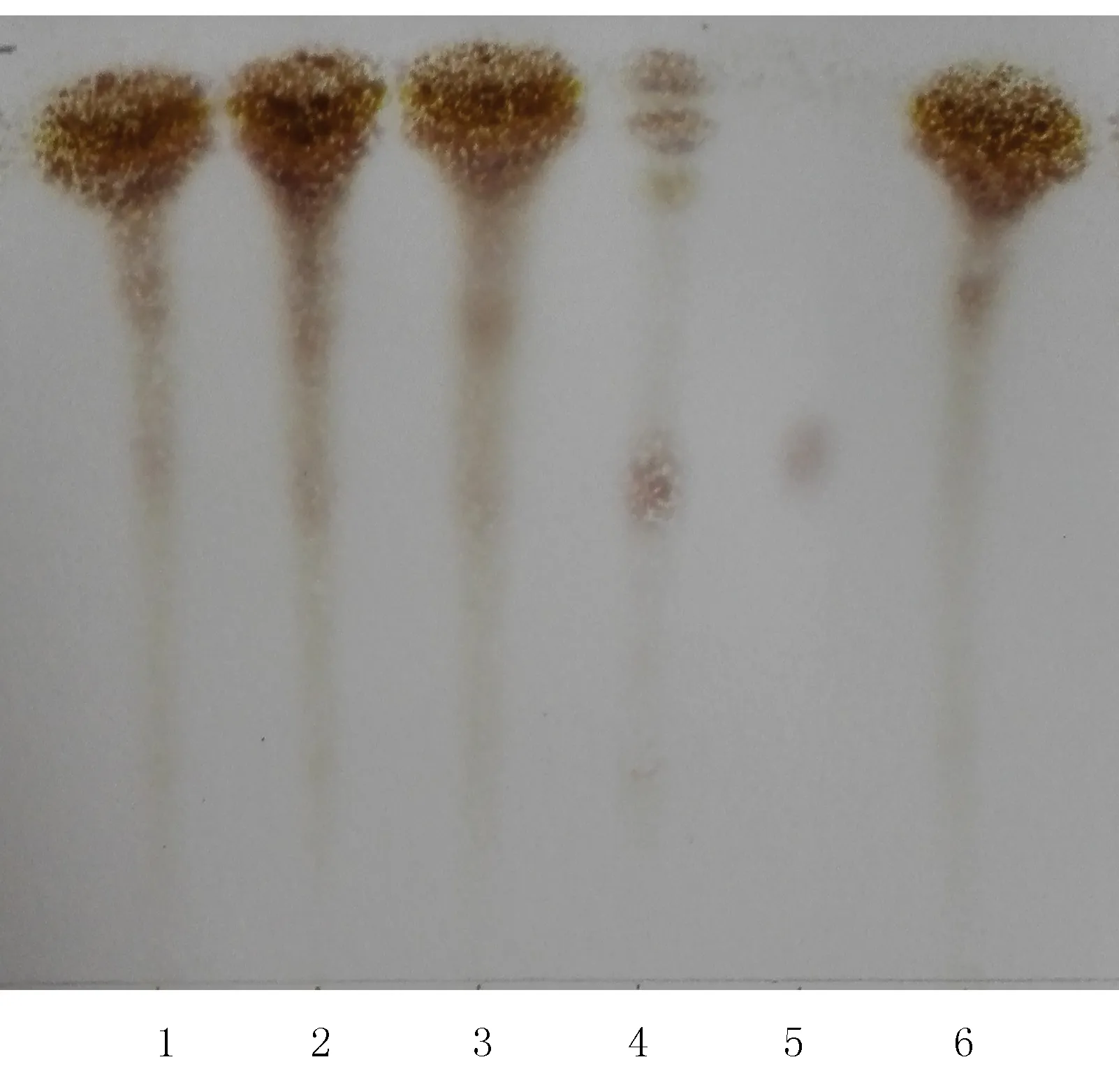
圖4 黃芪的薄層色譜圖
2.1.5 當歸的薄層鑒別[2]取復方參芪片,研細,稱取粉末10 g,加乙酸乙酯40 ml,超聲30 min,過濾,濾液蒸干,加乙醇1 ml溶解,作為供試品溶液。取缺當歸的復方參芪片陰性對照樣品10 g,同法制成陰性對照溶液。另取當歸對照藥材1 g,同法制成對照藥材溶液。吸取上述3種溶液各5 μl,分別點于同一以羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以正己烷-乙酸乙酯(4∶1)為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視。供試品色譜中,在與對照藥材色譜相應的位置上,顯相同的白色斑點,陰性對照無干擾。見圖5。
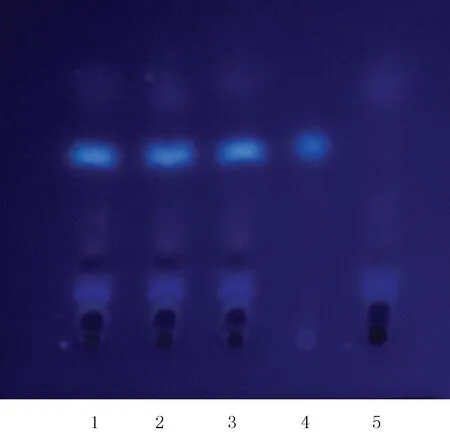
圖5 當歸的薄層色譜圖
2.2 補骨脂素和異補骨脂素的含量測定
2.2.1 色譜 條 件
Agilent XDB-C18 色譜柱 (250 mm×4.6 mm,5 μm),流動相為乙腈-水(34∶66),流速1.0 ml/min,檢測波長246 nm,柱溫25 ℃;進樣量為10 μl。理論塔板數按補骨脂素峰計算均≥3000。
2.2.2 對照品溶液的制備 精密稱取補骨脂素對照品11.32 mg和異補骨脂素對照品10.04 mg,分別置于100 ml容量瓶中,加甲醇溶解并定容,搖勻,分別作為對照品儲備液1(補骨脂素濃度為113.1 μg/ml )和對照品儲備液2(異補骨脂素濃度為100.4 μg/ml)。精密量取兩種對照品儲備液各10 ml,用甲醇定容至50 ml容量瓶,用甲醇稀釋至刻度,搖勻,作為混合對照品溶液(補骨脂素和異補骨脂素濃度分別為22.62和20.08 μg/ml)。
2.2.3 供試品溶液的制備 取復方參芪片樣品適量,研細,精密稱定0.2 g,置于具塞玻璃錐形瓶中,精密加入20 ml甲醇,稱定重量,超聲處理30 min,放冷,再次稱定重量,用甲醇補足缺失重量,搖勻,0.45 μm微孔濾膜過濾,取續濾液作為供試品溶液。
2.2.4 陰性對照溶液的制備 取缺補骨脂的陰性樣品粉末,按照2.2.3項下方法制備缺補骨脂的陰性對照溶液。
2.2.5 專屬性試驗 吸取對照品溶液、供試品溶液、陰性對照溶液各10 μl,分別按2.2.1項下色譜條件測定,記錄色譜峰。結果樣品中補骨脂素、異補骨脂素峰與其他組分色譜峰分離良好,陰性對照對檢測無干擾。見圖6。
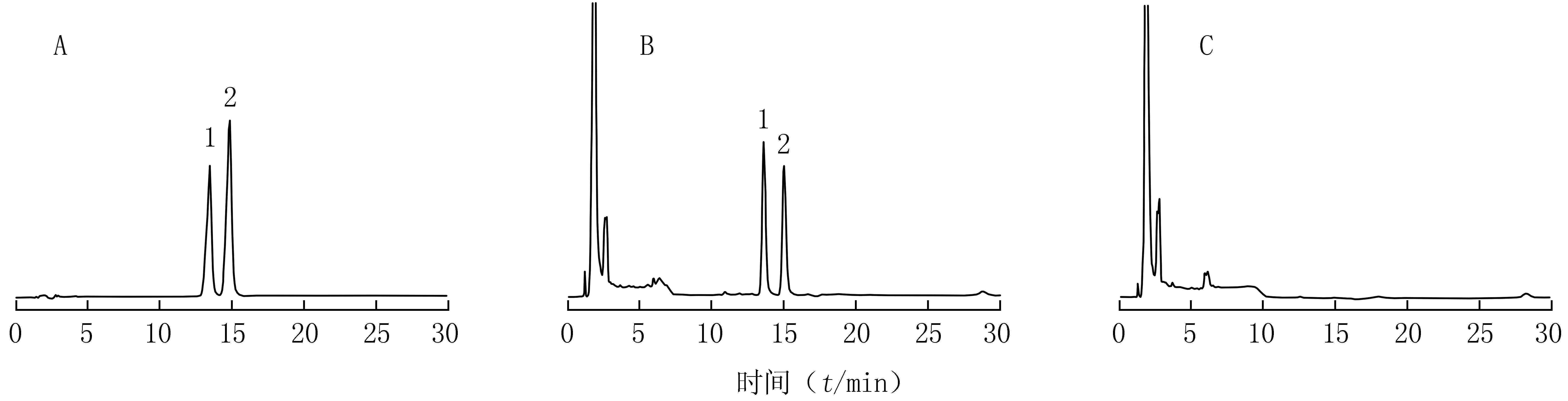
圖6 復方參芪片的HPLC譜圖
2.2.6 線性關系考察 精密吸取對照品儲備液1(補骨脂素濃度為113.1 μg/ml)和對照品儲備液2(異補骨脂素濃度為100.4 μg/ml)2.5、5.0、10.0、12.5、17.5、20.0、25.0 ml,分別用甲醇定容至50 ml容量瓶中,得濃度為5.65、11.31、22.62、28.27、39.58、45.23、56.54 μg/ml的系列補骨脂素對照品溶液和濃度為5.02、10.04、20.08、25.10、35.14、40.16、50.20 μg/ml的系列異補骨脂素對照品溶液。將上述不同濃度的補骨脂素對照品溶液和異補骨脂素對照品溶液分別注入色譜儀,進樣量為10 μl,記錄色譜峰。以峰面積(Y)為縱坐標,進樣濃度(X)為橫坐標,分別繪制標準曲線,得補骨脂素回歸方程為:Y=65.51X-6.817(r=0.999 9,n=7)。結果表明,補骨脂素在5.65~56.54 μg/ml范圍內有良好的線性關系。異補骨脂素回歸方程為:Y=70.27X+11.33(r=0.999 9,n=7)。結果表明,異補骨脂在5.02~50.20 μg/ml范圍內有良好的線性關系。
2.2.7 精密度試驗 取補骨脂素和異補骨脂素濃度分別為22.62和20.08 μg/ml的對照品溶液,精密吸取10 μl,注入色譜儀,重復進樣6次,測定其峰面積。結果補骨脂素峰面積的RSD=0.8%(n=6),異補骨脂素峰面積的RSD=0.9%(n=6),表明儀器精密度良好。取復方參芪片(批號20140421)供試品溶液,精密吸取10 μl,注入色譜儀,重復進樣6次,測定其峰面積,結果補骨脂素峰面積的RSD=0.8%(n=6),異補骨脂素峰面積的RSD=0.8%(n=6),表明該方法精密度良好。
2.2.8 穩定性試驗 取復方參芪片(批號20140421)供試品溶液,分別于0、2、4、8、10、12 h各進樣1次,每次10 μl,測定其峰面積,結果補骨脂素峰面積的RSD=0.8%(n=6),異補骨脂素峰面積的RSD=0.8%(n=6),表明復方參芪片供試品溶液至少在12 h內穩定。
2.2.9 加樣回收率試驗 取補骨脂素和異補骨脂素濃度分別為22.62和20.08 μg/ml的對照品溶液備用,取復方參芪片(批號20140421)適量,研細,精密稱取9份粉末,每份0.100 g,按2.2.3項下方法制成供試品溶液,分別按照供試品中含補骨脂素和異補骨脂素量的0.8倍、1倍、1.2倍加入對照品溶液,各平行做3份,并按2.2.1項色譜條件測定,根據測得量和加入量,計算各待測成分的加樣回收率。結果補骨脂素和異補骨脂素的平均加樣回收率分別為(99.80±1.8)%和(100.8±2.5)%(n=9)。
2.2.10 含量測定結果 分別取3個批次的復方參芪片適量,研細,每批次取粉末3份,每份0.2 g,按照2.2.3項下方法制備供試品溶液,測定含量,結果批號為20140421、20140910、20150317的復方參芪片中,補骨脂素的含量分別為(1.94±0.025)、(1.45±0.032)、(1.47±0.016) mg/g,異補骨脂素的含量分別為(1.65±0.020)、(1.35±0.022)、(1.38±0.015) mg/g(n=3)。
3 討 論
3.1 薄層鑒別的改進 本研究改進了原質量標準中補骨脂和丹參的鑒別方法,將補骨脂的鑒別方法改為TLC法,使得方法簡單易操作,并通過多次實驗,調整展開劑的比例,使薄層色譜顯色清晰。在丹參的薄層鑒別中,用二氯甲烷替代三氯甲烷作為展開劑,使得試劑的毒性降低。增加了原標準中沒有的柴胡、黃芪、當歸的薄層鑒別項,分別通過反復多次實驗調整展開劑的組成及比例,使得新增加的鑒別項專屬性強,所用試劑毒性小、配制簡單,且鑒別方法簡便易操作。
3.2 HPLC法吸收波長和流動相的選擇 根據參考文獻[2,7-9],選擇246 nm為最佳吸收波長。預實驗嘗試了不同的流動相,如甲醇-水(45∶55、50∶50、55∶45、52∶48、47∶53)、乙腈-水(47∶53),使用這些流動相時,待測成分與雜質峰均有干擾。當流動相為乙腈-水(34∶66)時,未有雜質峰干擾且出峰時間較短。
3.3 HPLC法提取溶劑用量的選擇 在供試品溶液的制備中,本研究分別考察了選用10、20、30 ml的甲醇作為提取溶劑量,并對所得結果使用SPSS 20.0軟件進行單因素方差分析,結果提取溶劑為10 ml 組與20 ml組的數據之間存在顯著性差異(P<0.05),20 ml 組與30 ml組之間不存在顯著性差異(P>0.05),為保證提取效果且節約溶劑用量,故選擇20 ml作為提取溶劑的用量。
3.4 HPLC法提取時間的選擇 在供試品溶液的制備時,本研究同時考察了3種不同提取時間的效果,分別為超聲處理(功率500 W,頻率40 kHz)20、30、40 min,同樣對3組結果使用SPSS 20.0軟件進行單因素方差分析,結果提取20 min組與提取30 min組的數據之間存在顯著性差異(P<0.05),提取30 min組與提取40 min組之間無統計學差異(P>0.05),為節約實驗時間,故選擇30 min作為提取時間。
本研究確立了復方參芪片中補骨脂素和異補骨脂素的含量測定方法,本方法簡便、有效、易操作,能夠有效地控制復方參芪片的質量,使本制劑的質量標準得到完善,并進一步保障了臨床的用藥安全性。
