晚期糖基化終末產物對糖尿病小鼠角膜樹突狀細胞活化的影響及作用機制
2021-07-23 06:33:00張雅妮白曉飛韋超
精準醫學雜志 2021年3期
張雅妮 白曉飛 韋超
(1 青島大學,山東 青島 266071; 2 山東第一醫科大學(山東省醫學科學院)山東省眼科研究所;3 山東第一醫科大學(山東省醫學科學院)山東省眼科研究所,山東省眼科學重點實驗室-省部共建國家重點實驗室培育基地)
晚期糖基化終末產物(AGEs)是由蛋白質、脂質以及核酸的游離氨基與還原糖的羰基發生糖基化反應所形成[1]。正常條件下,AGEs在體內的累積與年齡呈正相關[2]。糖尿病可導致AGEs在體內各組織內呈病理性蓄積,是引起糖尿病并發癥的重要原因之一[3-4]。目前,在糖尿病大鼠的角膜上皮、基質以及內皮層細胞均檢測到AGEs的沉積,而且AGEs的沉積與糖尿病角膜病變引起的上皮愈合延遲有關[5-6]。樹突狀細胞(DCs)作為一種強有效的抗原遞呈細胞,在控制炎癥反應、促進免疫耐受、募集免疫細胞和產生抗病毒細胞因子等生理過程中發揮重要作用[7]。在正常角膜基質及上皮層中均發現有DCs,呈角膜外周密集而角膜中央稀疏分布[8]。研究發現,DCs損耗可以延遲角膜上皮傷口愈合以及角膜神經的再生[9]。Toll樣受體4(TLR4)作為TLRs家族的成員,可上調炎性因子和干擾素的表達,引起炎癥和免疫反應[10-11]。已有相關研究顯示,TLR4信號通路參與了糖尿病引起的氧化應激和炎癥反應,而TLR4抑制劑瑞沙托維(TAK-242)可有效逆轉糖尿病引起的活性氧產生和核因子κB(NF-κB)活性[12-13]。然而,糖尿病條件下過度堆積的AGEs能否通過調控TLR4信號通路活化DCs尚不清楚。本研究旨在通過動物實驗和細胞實驗研究探討AGEs對糖尿病小鼠角膜中DCs活化功能的影響及相關機制。
1 材料與方法
1.1 材料和設備
SPF級雄性C57BL/6小鼠36只,購自北京維通利華實驗動物繁育有限公司,4~6周齡。1640培養基(美國HyClone公司),白細胞介素4(IL-4)、粒細胞-巨噬細胞集落刺激因子(GM-CSF)(美國PEPROTECH公司),TAK-242(美國MedChemExpress公司),SYBR Green qPCR試劑盒(南京諾唯贊生物科技股份有限公司),AGE-BSA、雙抗夾心ELISA試劑盒(美國Abcam公司)。
1.2 實驗方法
1.2.11型糖尿病小鼠模型的建立 選用6周齡小鼠,隨機分為正常對照組(A組)12只和1型糖尿病組(B組)18只。A組小鼠連續5 d腹腔注射0.1 mL檸檬酸-檸檬酸鈉緩沖液,B組小鼠連續5 d腹腔注射鏈脲佐菌素60 mg/kg。3個月后當小鼠血糖值>16.7 mmol/L,即為1型糖尿病小鼠建模成功。A、B組小鼠各6只,斷頸處死后取下眼球,剪下角膜,用于后續的Western Blot實驗。其他小鼠用于后續角膜上皮損傷模型構建。
1.2.2體外誘導小鼠骨髓源樹突狀細胞(BMDCs)4周齡正常小鼠6只,斷頸處死后分離股骨和脛骨,用1640培養基沖洗骨髓腔至其變白。制備為單細胞懸液后,加入紅細胞裂解液靜置3 min,離心棄上清液,PBS重懸2次以后。加入含10 μg/L IL-4和20 μg/L GM-CSF的1640培養基,重懸后接種。置于37 ℃、含體積分數0.05 CO2的恒溫箱中培養。培養3 d后更換為含有IL-4和GM-CSF(濃度同前)的1640培養基。培養6 d后用于后續實驗。
1.2.3小鼠BMDCs分組及處理 將培養6 d的BMDCs按實驗設計隨機分為C、D、E、F、G、H組。C、F組為空白對照組,于1640培養基中培養12 h;D組為牛血清白蛋白(BSA)組,于含有200 mg/L BSA的1640培養基中培養12 h;E、G組為AGE-BSA組,于含200 mg/L AGE-BSA的1640培養基中培養12 h;H組為TAK-242組,使用5 μmol/L TAK-242預處理1 h后,加入同G組的培養基培養12 h,然后進行后續試驗。
1.2.4酶聯免疫吸附法(ELISA)檢測各組細胞上清液內干擾素-β(IFN-β)蛋白的濃度 分別收集C、D、E、F、G、H組小鼠BMDCs上清液,嚴格按照ELISA試劑盒說明書操作,檢測各組細胞上清液中IFN-β蛋白的濃度。
1.2.5Western Blot實驗檢測各組小鼠的角膜內AGEs及各組細胞內p-IRF3、p-p65蛋白的表達水平 向A、B組角膜以及C、D、E、F、G、H組BMDCs內加入裂解液(含體積分數0.01磷酸酶抑制劑和體積分數0.01蛋白酶抑制劑)冰上裂解30 min,收集裂解液并超聲破碎,隨后離心吸取上清液,分別獲得角膜組織與細胞總蛋白,利用BCA試劑盒檢測蛋白濃度。將20 μg蛋白樣品上樣至含體積分數0.10 SDS-PAGE凝膠中,電泳、轉膜;用含體積分數0.05 BSA室溫封閉1 h,再分別加入一抗AGEs、干擾素調節因子3(IRF3)、磷酸化IRF3(p-IRF3)、p65以及p-p65,4 ℃孵育過夜,次日加入1∶5 000的辣根過氧化物酶標記的二抗,室溫孵育2 h;以蛋白印跡成像系統進行顯影。以GAPDH/β-actin作為對照,使用Image Lab軟件分析條帶灰度值。
1.2.6實時熒光定量PCR(RT-qPCR)檢測各組細胞炎性因子的表達 小鼠BMDCs按步驟1.2.2培養6 d后,按步驟1.2.3分為C、D、E、F、G、H組,6組細胞采用艾德萊試劑盒提取細胞內總RNA。然后根據諾唯贊反轉錄試劑盒說明書,在RNA內加入4×gDNA wiper Mix,42 ℃作用2 min;隨后加入5×HiScript Ⅲ qRT SuperMix,37 ℃作用15 min;繼以85 ℃作用5 s,然后反轉錄為cDNA。根據SYBR Green嵌合熒光法進行qPCR,以GAPDH作為內參照。引物由美國Invitrogen公司設計并合成,引物序列見表1。采用2-△△CT方法計算各組BMDGs內IFN-β、CXC基序趨化因子10(CXCL10)、白細胞介素-6(IL-6)和白細胞介素-12p35(IL-12p35) mRNA的相對表達量。

表1 小鼠關鍵基因引物核苷酸序列
1.2.7小鼠角膜上皮損傷模型構建及分組與處理將18只6周齡小鼠隨機分為I、J、K組,每組6只。I組在按照A組小鼠處理的基礎上于結膜下注射溶劑(體積分數0.01二甲基亞砜+體積分數0.99 PBS);J組在按照B組小鼠處理的基礎上于結膜下注射溶劑(同I組);K組小鼠在按照B組小鼠處理的基礎上于結膜下注射0.375 g/L TAK-242,TAK-242溶于溶劑內(同I組)。所有小鼠腹腔注射麻醉藥全身麻醉后置于操作臺上,以無齒鑷固定眼球,用直徑2.5 mm的環鉆輕壓中央角膜上皮層,輕輕旋轉環鉆在上皮層留下壓痕,不損傷角膜基質層,用上皮刮刀將壓痕范圍內的角膜上皮組織刮除,致角膜上皮組織損傷。將熒光素鈉染液滴在小鼠眼球表面,于刮除后0、24和40 h在裂隙燈下觀察小鼠角膜上皮修復情況并拍照。
1.3 統計學方法
2 結 果
2.1 兩組小鼠角膜內AGEs表達比較
Western Blot實驗檢測結果顯示,與A組小鼠相比,B組小鼠角膜組織中AGEs蛋白的水平顯著增高。見圖1。
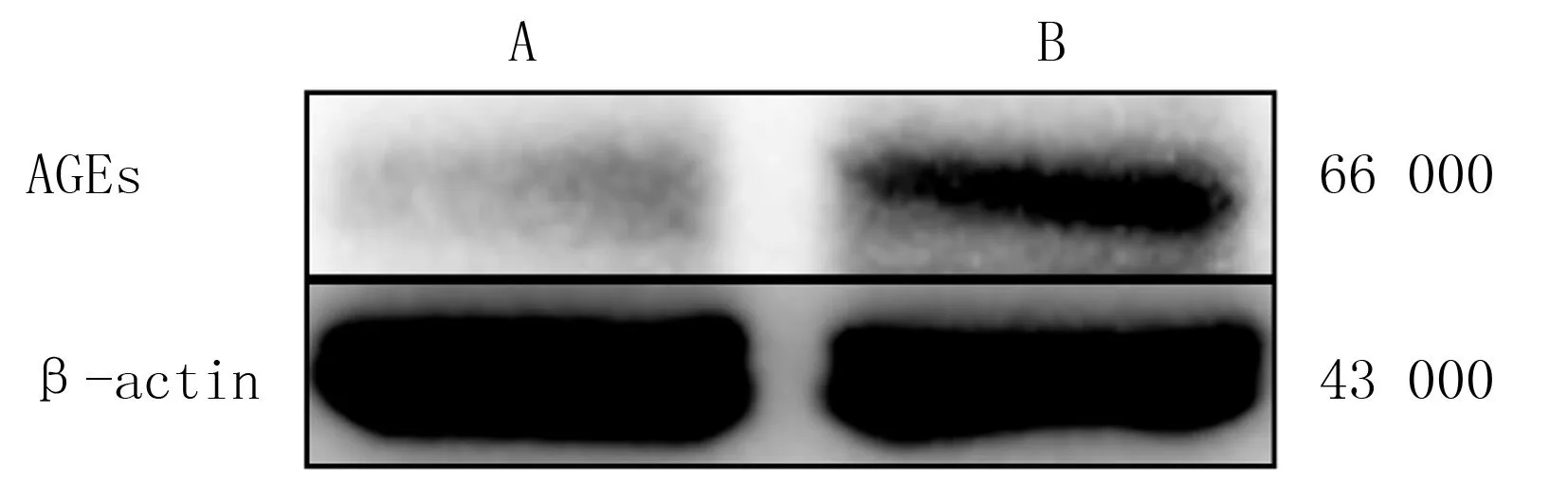
A、B分別對應A、B組圖1 兩組小鼠角膜組織AGEs蛋白表達情況
2.2 各組小鼠BMDCs活化情況比較
ELISA結果顯示,C、D、E組BMDCs上清液內IFN-β蛋白的表達量比較差異具有顯著意義(F=176.52,P<0.05),E組BMDCs上清液中IFN-β表達量顯著高于C、D組(t=13.51、14.95,P<0.05)。見表2。RT-qPCR分析顯示,3組小鼠BMDCs內IFN-β、CXCL10、IL-6和IL-12p35 mRNA相對表達量比較差異具有顯著意義(F=46.98~4 251.15,P<0.05),E組細胞內IFN-β、CXCL10、IL-6以及IL-12p35 mRNA的相對表達量均顯著高于C、D組(P<0.05)。見表2。Western Blot結果顯示,E組TLR4通路下游關鍵蛋白p-IRF3和p-p65的表達量較C、D組明顯增加。見圖2。
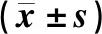
表2 3組小鼠BMDCs中炎性因子表達水平的比較
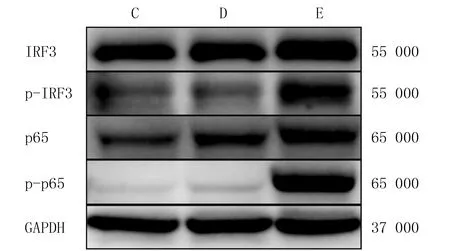
C、D、E分別為C、D、E組圖2 3組小鼠BMDCs中IRF3、p-IRF3、p65和p-p65蛋白的表達水平
2.3 TAK-242干預后各組小鼠BMDCs活化情況比較
ELISA檢測結果顯示,F、G、H組BMDCs上清液內IFN-β蛋白表達量比較差異具有顯著性(F=23.01,P<0.05),其中G組細胞上清液中IFN-β蛋白的表達量顯著高于F組(t=34.90,P<0.05),H組細胞上清液中IFN-β蛋白的表達量顯著低于G組(t=34.97,P<0.05)。見表3。RT-qPCR檢測結果顯示,3組小鼠BMDCs內IFN-β、CXCL10、IL-6和IL-12p35 mRNA相對表達量比較差異具有顯著性(F=44.64~1 257.10,P<0.05),其中G組細胞內IFN-β、CXCL10、IL-6和IL-12p35 mRNA相對表達量顯著高于F組(P<0.05),H組低于G組(P<0.05)。見表3。Western Blot實驗結果顯示,H組TLR4通路下游相關蛋白p-IRF3和p-p65表達水平較G組明顯降低。見圖3。
表3 3組小鼠BMDCs中炎性因子表達水平的比較
2.4 TAK-242對糖尿病小鼠角膜上皮損傷修復的影響
組別、時間及其交互作用均對小鼠角膜上皮損傷面積具有明顯影響(F組別=19.94,F時間=446.68,F交互=9.96,P<0.05)。單獨效應結果顯示,在24、40 h,3組小鼠角膜上皮損傷面積比較差異有統計學意義(F=5.85、4.89,P<0.05),其中J組角膜上皮損傷面積顯著高于I組(P<0.05),K組低于J組(P<0.05)。3組小鼠不同時間點角膜上皮損傷面積比較差異有顯著性(F=8.97~16.69,P<0.05),其中,24 h上皮損傷面積均小于0 h(P<0.05),40 h上皮損傷面積均小于24 h(P<0.05)。見圖4、表4。
表4 各組小鼠角膜上皮刮除后上皮損傷面積比較
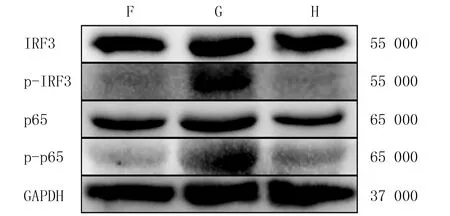
F、G、H分別為F、G、H組圖3 3組小鼠BMDCs中IRF3、p-IRF3、p65和p-p65蛋白的表達水平
3 討 論
AGEs作為一類復雜的非酶促蛋白質翻譯后修飾產物,參與了糖尿病角膜病變、糖尿病腎病、糖尿病足及糖尿病視網膜病變等多種糖尿病并發癥的發病過程[5,14-16]。AGEs在角膜上皮基底膜的沉積可影響上皮細胞與基底膜的黏附,進而引發糖尿病角膜上皮的改變,是引起糖尿病角膜病變的重要機制之一[17]。本實驗通過Western Blot實驗檢測顯示,糖尿病小鼠角膜表達AGEs蛋白較正常小鼠明顯增多,進一步證實了AGEs在糖尿病角膜病變中異常沉積。
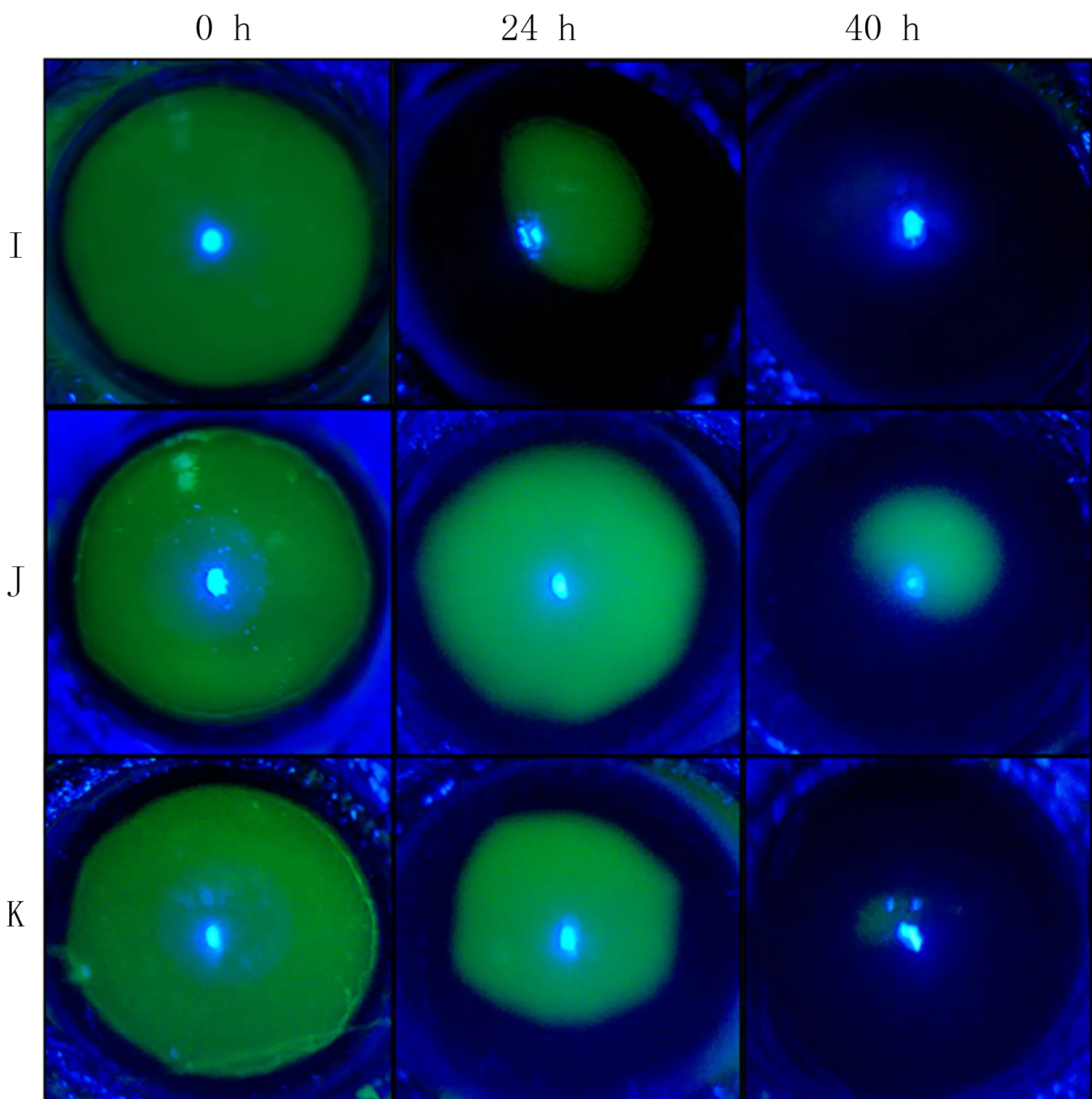
I、J、K分別對應I、J、K組圖4 各組小鼠角膜上皮刮除后熒光素鈉染色結果
近年來,LEPPIN等[18]發現DCs在糖尿病及其并發癥的致病過程中發揮關鍵性作用,糖尿病角膜病變中角膜DCs的增加可能與神經纖維減少存在關聯。研究表明,DCs可以通過模式識別受體(例如Toll樣受體)識別外來物從而起到了保護角膜的作用[19]。TLR4作為Toll樣受體家族成員,已被證實參與了糖尿病的發病過程[20]。TLR4信號通路主要通過MyD88/NF-κB和TRIF/IRF3兩條途徑,引起下游信號級聯反應以及誘發慢性炎癥反應,該炎癥反應是糖尿病角膜病變發生發展的關鍵因素之一[21-22]。TLR4/TRIF/IRF3途徑主要參與Ⅰ型干擾素的表達[23]。糖尿病通過活化TLR4/NF-κB加劇氧化應激、炎癥反應,進而參與到糖尿病并發癥的發病過程中[24-25]。基于角膜內DCs數量少且難分離,本研究通過體外原代培養BMDCs模型后發現,AGE-BSA可引起BMDCs內p-p65和p-IRF3蛋白表達增加,這表明AGE-BSA可能通過影響TLR4/NF-κB和TLR4/IRF3兩條途徑來調控BMDCs的活化,進一步上調下游炎性相關細胞因子的表達(包括IFN-β、CXCL10、IL-6和IL-12p35),進而參與到糖尿病角膜病變的慢性炎癥反應中。TAK-242作為一種選擇性TLR4抑制劑,可直接結合在TLR4胞內結構域中抑制其活化[26]。本實驗結果顯示,TAK-242干預后,BMDCs內p-p65和p-IRF3蛋白的表達均降低,且通路下游的炎性相關細胞因子水平亦顯著下調,提示阻斷TLR4信號通路可以抑制AGE-BSA對于BMDCs的活化作用,進一步表明AGE-BSA可能通過TLR4信號通路調控BMDCs的活化。
糖尿病患者在遭受眼部創傷或手術后易發生淺表點狀角膜炎、復發性角膜糜爛及持續性上皮缺損等癥狀[27]。糖尿病角膜病變引起的病理改變包括AGEs的沉積、角膜上皮基底膜成分的改變、角膜神經的損傷以及氧化應激等[28]。AGEs的累積可觸發細胞凋亡、氧化應激及慢性炎癥等病理過程,進一步加重糖尿病患者角膜的病變[29-30]。基于此,本研究旨在探討TLR4信號通路在糖尿病角膜上皮損傷修復過程中的作用。熒光素鈉染色結果顯示,在角膜上皮刮除后24、40 h,糖尿病小鼠角膜上皮愈合較正常小鼠延遲,與之前報道的結果一致[31]。而糖尿病小鼠結膜下注射TAK-242干預后,上皮修復速度較糖尿病對照小鼠加快,說明TLR4信號通路可能參與了糖尿病角膜上皮損傷修復延遲的病理過程。
綜上所述,本研究證實糖尿病小鼠角膜內存在AGEs過表達,AGE-BSA可能通過調控TLR4信號通路來促進BMDCs分泌炎性相關細胞因子,TLR4信號通路可能參與了糖尿病小鼠角膜上皮損傷修復延遲愈合的過程。本研究不足在于角膜內DCs含量比較少,在動物水平驗證其功能變化存在一定困難,僅能通過其對角膜上皮愈合的影響間接驗證,后續研究將對此不足進一步完善。本研究結果為揭示TLR4信號通路在糖尿病角膜病變防治中的作用提供了實驗依據。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年11期)2021-08-22 03:15:16
學苑創造·A版(2020年9期)2020-10-13 09:41:02
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
